---
title: 'Making bio.tools fit for workflows'
title_short: 'Making bio.tools fit for workflows'
tags:
- scientific workflows
- computational pipelines
- bio.tools
- EDAM
- APE
authors:
- name: Anna-Lena Lamprecht
orcid: 0000-0003-1953-5606
affiliation: 1
- name: Veit Schwämmle
orcid: 0000-0002-9708-6722
affiliation: 2
- name: Vedran Kasalica
orcid: 0000-0002-0097-1056
affiliation: 1
- name: Mads Kierkegaard
orcid: 0000-0001-5651-6215
affiliation: 2
- name: Hans Ienasescu
affiliation: 3
- name: Magnus Palmblad
orcid: 0000-0002-5865-8994
affiliation: 4
affiliations:
- name: Dept. of Information and Computing Sciences, Utrecht University, Netherlands
index: 1
- name: Dept. of Biochemistry and Molecular Biology, University of Southern Denmark, Denmark
index: 2
- name: National Life Science Supercomputing Center, Technical University of Denmark, Denmark
index: 3
- name: Leids Universitair Medisch Centrum: Leiden, Zuid-Holland, Netherlands
index: 4
date: 9 November 2021
bibliography: paper.bib
event: BioHackathon Europe 2021
biohackathon_name: "BioHackathon Europe 2021"
biohackathon_url: "https://biohackathon-europe.org/"
biohackathon_location: "Barcelona, Spain, 2021"
group: 22
# URL to project git repo --- should contain paper.md
git_url: https://github.com/bio-tools/biohackathon2021/
# This is the short authors description that is used at the
# bottom of the generated paper.
authors_short: TBD \emph{et al.}
---
With 20.000+ entries, bio.tools is a major registry of computational tools in the life sciences. During the 2021 European BioHackathon, the project "Making bio.tools fit for workflows" was run to address two urgent needs of the platform:
-
Slicing the bio.tools content through specialisation and categorisation, to improve exposure to communities and to present useful content for the users. The main challenge is to summarise relevant information from the wealth of annotation categories in bio.tools and metrics from external sources. Therefore we aim to enrich tools, communities and collections with statistics and metrics that summarise functionality, impact and annotation quality. These metrics and statistics are valuable resources for tool-building communities, scientific domains, individual scientific tool repositories and groups specialising in technical features. With that information, we can identify, calculate and integrate metrics relevant for the bio.tools registry. In addition we will devise a mock-up / alpha version summary stats page within bio.tools.
-
Improving the quality of functional tool annotations, to enable automated composition of individual tools into multi-step computational pipelines or workflows. Currently, tool annotations are often incomplete or imprecise, hampering plug&play workflow composition. We will develop a protocol for improving functional tool annotations in bio.tools. It will be based on 1) selecting reference workflows from workflow repositories and literature, 2) trying to recreate them using bio.tools and the Automated Pipeline Explorer, 3) comparing automatically created and reference workflows, and 4) if necessary revising the tool annotations until recreation succeeds. Workshop participants will perform this process and concurrently develop the tooling and documentation to enable its application to additional workflows after the hackathon.
The outcomes of this project will make software more findable and provide a solid basis for iteratively improving the quality of functional annotations in bio.tools, making it an increasingly powerful source of new fit-for-purpose workflows.
In this document we document the progress towards these goals that was made during the BioHackathon
(please add content here)
General trends and properties within a tool collection. The main use case discussed here consists of the tools withe an EDAM topic Proteomics.
Word clouds provide an overview of the different terms and their frequency.
Differences between input and output data:

as well as between input and output formats:

We can monitor statistics on tool type, operation system and license

Availability of code monitored by OpenEBench:

More stats from Europe PMC, counting citations of the main publications of the tools

Here, red is the distribution of about 50% of all bio.tools tools, grey the publications of proteomics tools.
In order to asses the quality of bio.tools annotation, we have decided to distinguish few different aspects of evaluation:
How well are the tools annotated in comparison with all annotations in bio.tools (red: proteomics tools, grey: all):

In general, proteomics have more EDAM annotations (also counts for Data and Format).
One of the straight-forward criteria for accessing the quality of annotation is distinguishing tools that include input/output pairs, and those that do not. We observe that ~7,6% (1721/22540) of the tools provide the input/output annotations with both, data type and format specified, while the others lack at least one of the mentioned annotations. In addition, we can see that the percentage changes within different domains,/topics, which does not come as a surprise. Where the domains such as 'proteomics' include ~36,4% (273/751) of the fully annotated tools, with respect to the input/output annotations.
(to add figure)
When looking at tools that have annotated inputs and outputs, we try to asses the "IO concreteness" of tool annotations by evaluating how specific the terms used to specify the inputs and outputs are, i.e., whether the terms are abstract classes - low "IO concreteness", or concrete concepts (usually leaves) - high "IO concreteness". We noticed that overall "IO concreteness" of inputs is slightly higher than the outputs, i,e., the terms used for input annotations are more specific/concrete than the terms used for output. Furthermore, the data formats of both, inputs and outputs, have higher "IO concreteness" than data types. As a matter of fact over 20% of the input/output annotations are annotated as Data, which is the root term of the corresponding taxonomy.
(to add figure maybe)
The results of the evaluation of the "IO concreteness" of bio.tools suggest that there is space for improvement when to comes to data types annotations of the operation inputs/outputs.
We use existing EDAM object properties to attempt to automatically improve the corresponding bio.tools annotations. We identify three key object properties, as follows:
- is_format_of - defines dependency between data formats and corresponding data types
- has_input - defines dependency between operation and corresponding data type inputs
- has_output - defines dependency between operation and corresponding data type outputs
We focus on the is_format_of as part of the preliminary approach, with plan to extend it to all 3 concepts. The results show that out of 1721 operations, there are 443 inputs and outputs, where data type suggested by EDAM ontology (based on the given format and is_format_of object property) is more concrete than the one annotated in bio.tools (see file). When looking at the proteomics domain, there are 16 inputs and outputs (out of 273 operations) where data type suggested by EDAM ontology (based on the given format and is_format_of object property) is more concrete than the one annotated in bio.tools (see file).
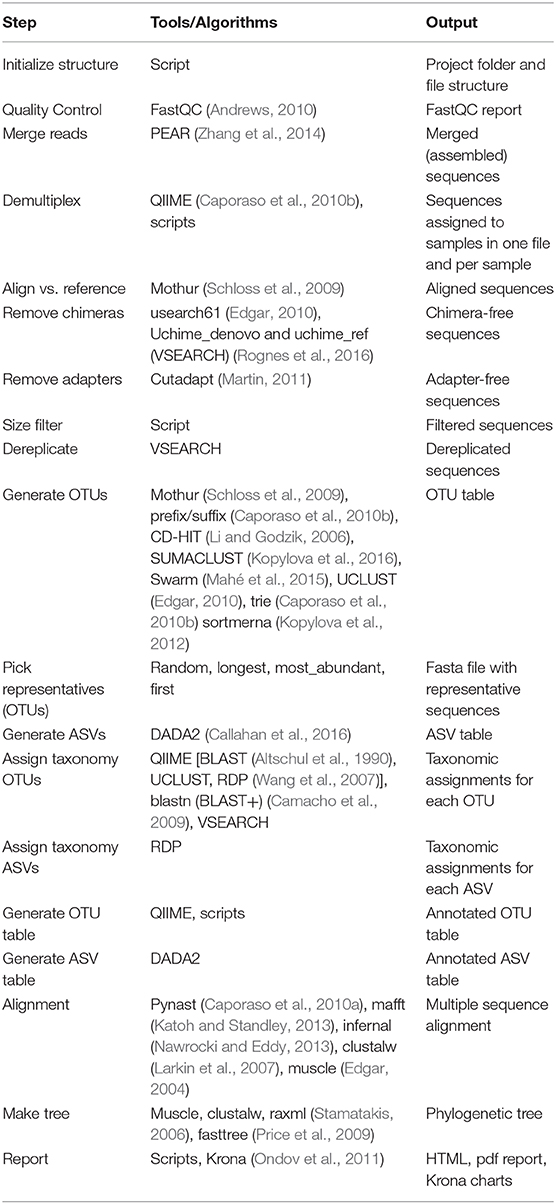
Cascabel [@cascabel] is a variable pipeline for amplicon sequence data analysis, developed by the department of Marine Microbiology and Biogeochemistry at NIOZ Royal Netherlands Institute for Sea Research in collaboration with the department of Earth Sciences at Utrecht University, Netherlands.
It makes an interesting use case for for this project, as it already foresees several variants of the main workflow, and potentially more can be found when taking to account the full content of bio.tools.
The tools used in Cascabel are listed in the figure/table at https://www.frontiersin.org/files/Articles/489357/fgene-11-489357-HTML/image_m/fgene-11-489357-t001.jpg
{kind=link}
We start with a first assessment of the status of the tools listed in bio.tools, summarized in the table below. Note that the custom scripts mentioned are obviously not available in bio.tools (here's the shim discussion again!) and therefore not included in the table.
| Tool | in bio.tools? | bio.tools ID | functional annotation? |
|---|---|---|---|
| FastQC | yes | biotools:fastqc | yes |
| PEAR | yes | biotools:pear | no |
| QIIME | yes | biotools:qiime | no |
| QIIME | yes | biotools:qiime2 | no |
| Mothur | no | - | - |
| usearch61 | no | - | - |
| VSEARCH | yes | biotools:vsearch | yes |
| Cutadapt | yes | biotools:cutadapt | yes |
| Cutadapt 1.12 | yes | biotools:cutadapt_1.12 | no |
| CD-HIT | yes | biotools:cd-hit | yes |
| SUMACLUST | no | - | - |
| Swarm | yes | biotools:swarm | yes |
| UCLUST | no | - | - |
| trie | no | - | - |
| SortMeRna | yes | biotools:sortmerna | no |
| DADA2 | yes | biotools:dada2 | no |
| BLAST | yes | biotools:blast | no |
| pynast | no | - | - |
| MAFFT | yes | biotools:MAFFT | yes |
| Infernal | yes | biotools:infernal | yes |
| ClustalW | yes | biotools:clustalw | yes |
| MUSCLE | yes | biotools:muscle | yes |
| RAxML | yes | biotools:raxml | no |
| FastTree | yes | biotools:fasttree | no |
| Krona | yes | biotools:krona | no |
Next steps:
- add the missing tools to bio.tools
- check and if necessary improve the functional annotations
- identify additional tools in bio.tools with similar functionality and add them to the Cascabel collection/domain
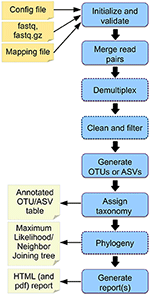
Further ahead, the goal is to create new variants of the Cascabel pipeline, ideally using automated workflow exploration as facilitated by APE [@apetool]. In addition to a well-annotated collection of tools, this needs suitable workflow specifications. These consist of available input data types and formats, and desired outputs types and formats, and can be supplemented with constraints expressing, e.g., operations to include or to avoid. These specifications are formulated using terms from the EDAM ontology.
The Cascabel paper includes a conceptual overview of the workflow at https://www.frontiersin.org/files/Articles/489357/fgene-11-489357-HTML/image_t/fgene-11-489357-g002.gif, which can serve as a blueprint for the required specification, pending its translation to adequate EDAM terms.
{kind=link}
....
We thank the organizers of the BioHackathon Europe 2021 for travel support for some of the authors.