anvi'o v6, "esther"
We are happy to announce a new version of anvi'o, "esther" (easy install through conda, quick try via docker)).
After nearly 9,000 changes that introduced about 16,000 new lines of code, the current version of anvi'o represents many fixes to big and small bugs, as well as new features. This page intends to give you a summary of most notable changes that come with esther.

The codename is a small tribute to Esther Lederberg (1922-2006), an American microbiologist who studied plasmids and bacterial viruses. Lederberg discovered lambda phage, an E. coli virus that is commonly used in bacterial genetics and molecular biology to deliver DNA into a recipient organism. This led to her description of specialized transduction, that occurs when a prophage improperly excises from the host chromosome carrying host DNA in addition to the viral DNA. In collaboration with her husband, Lederberg developed the technique known as replica plating, which allows repeatable inoculation of bacterial colonies. Lederberg and Luigi L. Cavalli-Sforza discovered the Fertility factor or F-plasmid in E. coli. This is a sequence of DNA that lets the host cell transfer genetic material via a rod-like structure into recipient cells (conjugation). Despite her many incredible scientific accomplishments, she was constantly overshadowed by her husband. She was not appointed to a tenured position while they were both faculty at Stanford, and after their divorce she had a difficult time retaining her appointment. We dedicate anvi'o version 6 to the memory and revolutionary discoveries of Dr. Lederberg.
Real-time estimation of genome taxonomy
Working with genomes often requires insights into their taxonomy. This becomes a critical need especially in genome-resolved metagenomics studies as we are burning to find out where the genomes we reconstruct from metagenomes fit in the tree of life. Until this esther, anvi’o did not offer anything to address this need, however, this new version comes with a novel solution that covers both the interactive interface during binning:

and the terminal environment to survey existing collections of genomes:
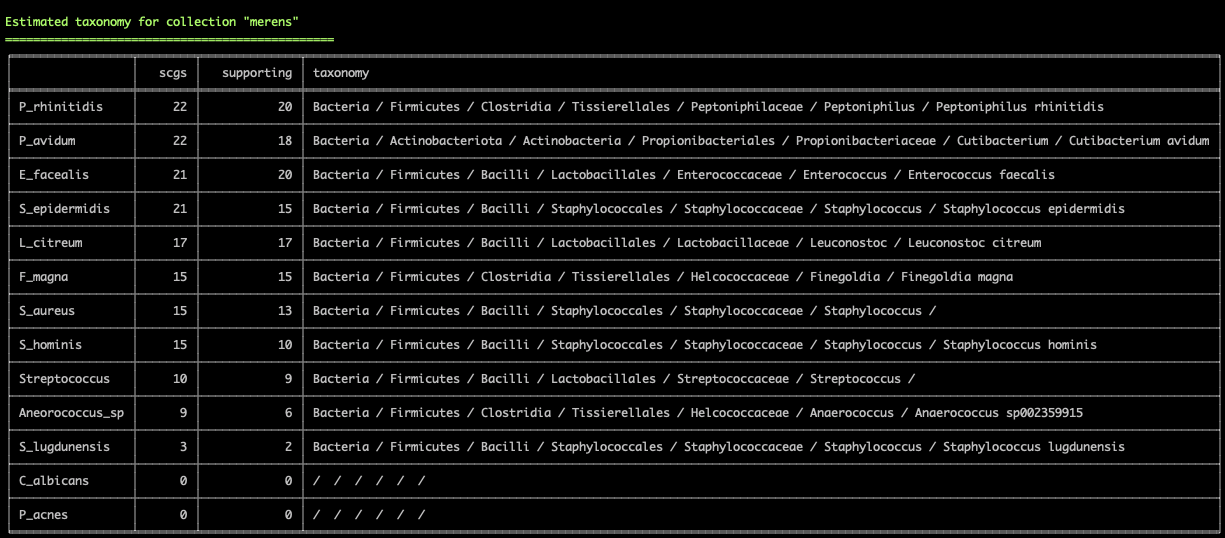
These two examples are from the infant gut dataset by Sharon et al (2013), which we often use to demonstrate anvi'o features, but we can't wait to hear from you to learn about your experience with this feature.
Please read in this article the usage details, our thanks to The Genome Taxonomy Database for making their raw data public, and potential caveats of our approach:
http://merenlab.org/scg-taxonomy
None of this would have been possible without the coding help from Quentin Clayssen and Özcan Esen, and critical suggestions from Alon Shaiber.
A new tool for genome de-replication
De-replication is a critical need to minimize bias in metagenomic read recruitment analyses. In our previous studies we had performed de-replication with a series of Python scripts, but no more. Thanks to Mahmoud Yousef and Evan Kiefl's efforts, we now have two new programs, anvi-compute-genome-similarity and anvi-dereplicate-genomes, integrated with metagenomic and pangenomic workflows in anvi'o and use sourmash and PyANI in the backend.
A tutorial for their usage is on the way!
Support for more binning algorithms
In previous versions of anvi'o we had a native module for CONCOCT, one of the popular binning algorithms for automatic clustering of contigs into genome bins. We have changed that behavior in this version. You will still be able to use the program anvi-import-collection to import binning results from ANY binning software as before, but anvi'o will also be able to automatically use binning tools existing on your system through our new program anvi-cluster-contigs. Here is a command line output to give you a sense of it:

This framework is highly modular, so the integration of new binning algorithms is extremely straightforward thanks to Özcan Esen's excellent design. If you are a programmer you can take a look at the module for MaxBin2 or BinSanity to develop one for your algorithm for benchmarking or testing efforts.
Effective ways to inspect and visualize contig coverages
Recognizing the importance of actually 'looking' at data, we have been putting a lot of emphasis on the inspection capabilities of anvi'o. When it comes to metagenomic read recruitment and coverages, inspecting contigs can be critical to gain deeper insights into what is actually going on.
In this version we have two new programs. The first one is anvi-inspect. The inspect page of anvi’o is very useful for careful examination of contig coverages and single nucleotide variants. Sometimes this might even be all you want. This new program enables you to immediately pull up the inspection page of a given contig without going through the whole hassle of opening the interactive interface.
We often feel the need to put coverage patterns of contigs in presentations or publications. Yet it becomes challenging when there are too many samples in a dataset as it makes it harder to study or save patterns comfortably using the interface. So we thought it would have been very useful if anvi'o could export coverage statistics using ggplot, but we didn't know enough R to be able to do this properly. As a result, we did what anyone who wish to work with talented people would do --we asked for help on Twitter:

Our call for help was heard by Ryan Moore, who actually developed a new anvi'o program that did exaxtly what we thought you would need, and much more: anvi-script-visualize-split-coverages (we sent him an anvi'o t-shirt as a token of our deep gratitude for his contribution, but we never got a photo back, so we don't know whether he is wearing it).
This program can export split coverages along with single-nuleotide variants on them into PDF files for even very large numbers of samples. It uses the output files anvi'o generates through anvi-get-split-coverages and optionally anvi-gen-variability-profile. The output is customizable with respect to plot color, axes, SNV color and grouping of samples. The tutorial for this feature will soon be on our web page.
Improved genome completion/redundancy estimates
New single-copy core gene collections
Starting with this version, we no longer use Campbell et al. and Rinke et al. single-copy core gene (SCGs) HMM sets to estimate completion of bacterial and archaeal genomes. Instead, we are using a modified version of the bacterial single-copy core gene collections Mike Lee recently described, and a set of BUSCO HMMs Tom Delmont curated. Now anvi'o can estimate the completion of bacterial, archaeal, and protist genomes (#1150).
New random forest domain of life classifier
In previous versions anvi'o has relied on multiple heuristics to predict the domains of selected contigs or genomes for the determination of which SCG collection to use to estimate and display completion and redundancy. In this version we have a brand new random forest classifier to take care of this challenging task. This robust classifier with appropriate addition of noise solves this issue like magic, and when you have a bunch of genomes, it gives you proper estimates in the interface (the example is also from the infant gut dataset),
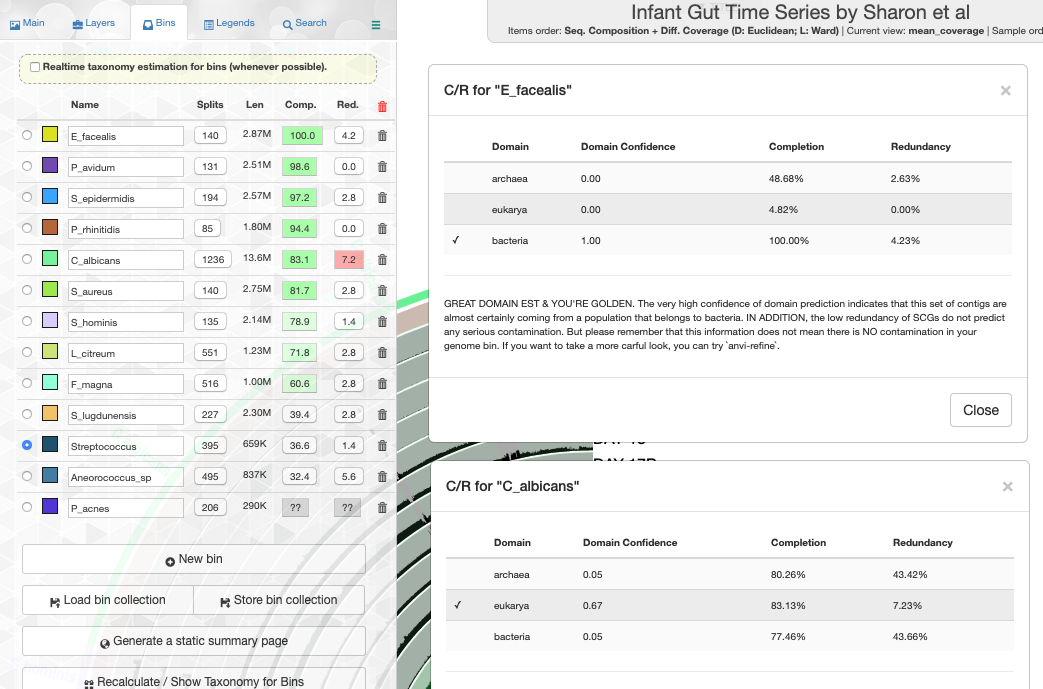
or in the terminal,

Undo/Redo for the interactive interface
Yes. This feature is finally here. Now when you make a mistake while curating or refining your genomes using anvi-interactive or anvi-refine, you will be able to use Ctrl + Z and Ctrl + Shift + Z key combinations for undo and redo your binning decisions. If you can't contain your emotions, consider taking Özcan Esen for a coffee for this excellent feature :)
A new tool to extract target loci from genomes and metagenomes
Some genetic analyses call for the comparison of specific genetic loci between genomes. For example, one may be interested in investigating evidence for adaptive evolution of the lac operon between different E. coli strains by extracting all loci from different genomes. Anvi'o esther comes with a very talented tool, anvi-export-locus, that will help you extract target loci from a larger genomic context, whether those context are genomes or metagenomic assemblies.
This tool cuts out loci using two approaches: default mode or what we call flank-mode. In the default mode, the tool locates a designated anchor gene, then cuts upstream and downstream context based on user-defined input. Flank-mode, on the other hand, locates designated genes that surround the target locus, then cuts in between them. Target genes of interest to locate anchors for exicion can be defined through their specific ids in anvi'o or through search-terms that query functional annotations or HMM hits stored in your contigs databse!
If you find it useful for your reserach, you can send post card to Alon Shaiber, Evan Kiefl, and the newest member of anvi'o developers, Matthew Schechter. A tutorial is on the way :)
Much faster HMMs
You complained, we heard (hehe). In anvi'o esther we finally fixed the sluggish speeds of HMM operations from which we you have suffered even when you assigned multiple threads to anvi-run-hmms. Özcan Esen revamped our code and has improved our speed dramatically with increasing number of threads given to anvi'o. Our tests indicate that speed gains roam around as much as four gazillion.
Much better functional enrichment analyses for pangenomes
Anvi'o esther comes with a new version of anvi-get-enriched-functions-per-pan-group thanks to the invaluable statistics input and code we have received from Amy Willis (@AmyDWillis). Please take a look at our tutorial on pangenomics for details.
Anvi'o gets better at helping you
Getting offline help from anvi'o has been difficult. Recognizing this limitation, Evan Kiefl created the program anvi-help that will help you find your way through anvi'o by simply asking anvi'o what does it have to do X. Here is an example. You type the following,
anvi-help functions
And you get back this:

As a part of our efforts to make information more accessible to you, Iva Veseli created a new resource: Getting help from the anvi'o community.
Thanks!
During the last few months the list of anvi'o developers grew rapidly, for which we are extremely grateful. The sixth version of the platform, which is now close to 80,000 lines of fully open Python and JavaScript code, would not have been possible without those who took their time to participate this community effort with their ideas and expertise.
We are very also very thankful for our users, whose feature requests, bug reports, and patience continue to give us energy to push things forward (although I can promise that we are not going to be pushing anything anywhere for a week or two after this release as we all just want to take a very long nap).
Finally, we thank all the open-source software developers and data curators everywhere. Without them none of these would have ever existed.
We hope esther helps you with your research 😇
To read the updated installation instructions for v6, please visit http://merenlab.org/install-anvio
If you are interested in anvi'o but don't know where to start, please read our "getting help" document, catch us in one of our free workshops, or find us on our Slack channel.