404
+ +Page not found
+ + +Page not found
+ + +The pipeline is done as part of MNI projects, it is written by Saeid Amiri with associate with Dan Spiegelman, and Sali Farhan at Neuro Bioinformatics Core. Copyright belongs MNI BIOINFO CORE.
+ +The output of finalseg.csv can be categorized to 1) locus and alleles, 2) CSQ, 3) Global- Non-Affected 4) Global-Affected, 5) Family, 6) Family-Affected 7) Family - Non-affected. If you do not want to have CSQ in the output file, choose CSQ=False.
locus: chromosome
+alleles: a variant form of a gene
VEP put all the requested information in infront CSQ.
+glb_naf_wild: Global - Non-Affecteds, wildtype
+glb_naf_ncl: Global - Non-Affecteds, no call
+glb_naf_vrt: Global - Non-Affecteds, with variant
+glb_naf_homv: Global - Non-Affecteds, homozygous for ALT allele
+glb_naf_altaf: Global - Non-Affecteds, ALT allele frequency
glb_aff_wild: Global - Affecteds, wildtype
+glb_aff_ncl: Global - Affecteds, no call
+glb_aff_vrt: Global - Affecteds, with variant
+glb_aff_homv: Global - Affecteds, homozygous for ALT allele
+glb_aff_altaf: Global - Affecteds, ALT allele frequency
{famid}_wild: Family - Affecteds: wildtype
+{famid}_ncl: Family - Affecteds: no call
+{famid}_vrt: Family - Affecteds: with variant
+{famid}_homv: Family - Affecteds: homozygous for ALT allele
{famid}_aff_wild: Family - Affecteds: wildtype
+{famid}_aff_ncl: Family - Affecteds: no call
+{famid}_aff_vrt: Family - Affecteds: with variant
+{famid}_aff_homv: Family - Affecteds: homozygous for ALT allele
{famid}_wild_naf: Family - Nonaffecteds: wildtype
+{famid}_ncl_naf: Family - Nonaffecteds: no call
+{famid}_vrt_naf: Family - Nonaffecteds: with variant
+{famid}_homv_naf: Family - Nonaffecteds: homozygous for ALT allele
MIT License
+Copyright (c) 2022 The Neuro Bioinformatics Core
+Permission is hereby granted, free of charge, to any person obtaining a copy +of this software and associated documentation files (the "Software"), to deal +in the Software without restriction, including without limitation the rights +to use, copy, modify, merge, publish, distribute, sublicense, and/or sell +copies of the Software, and to permit persons to whom the Software is +furnished to do so, subject to the following conditions:
+The above copyright notice and this permission notice shall be included in all +copies or substantial portions of the Software.
+THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR +IMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, +FITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE +AUTHORS OR COPYRIGHT HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER +LIABILITY, WHETHER IN AN ACTION OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, +OUT OF OR IN CONNECTION WITH THE SOFTWARE OR THE USE OR OTHER DEALINGS IN THE +SOFTWARE.
+ +This is the initial release.
+Supposed to
+The pipeline can be executed using a Singularity container, eliminating the need to install modules within the pipeline. It is compatible for use on High-Performance Computing (HPC) systems or any standard workstation linux system.
+Download the segregation image.
+singularity pull library://saeidamiri1/mni/seg_cont.sif:latest
+Execute singularity exec ./seg_cont.sif launch_segpy.sh -h to ensure the pipeline is functioning correctly.
------------------------------------
+Segregation pipline version 0.2.2.3
+
+Usage: /usr/local/bin/launch_segpy.sh [arguments]
+ mandatory arguments:
+ -d (--dir) = Working directory (where all the outputs will be printed) (give full path)
+ -s (--steps) = Specify what steps, e.g., 2 to run just step 2, 1-3 (run steps 1 through 3). 'ALL' to run all steps.
+ steps:
+ 0: initial setup
+ 1: create hail matrix
+ 2: run segregation
+ 3: final cleanup and formatting
+
+ optional arguments:
+ -h (--help) = Get the program options and exit.
+ --parser = 'general': to general parsing, 'unique': drop multiplicities
+ -v (--vcf) = VCF file (mandatory for steps 1-3)
+ -p (--ped) = PED file (mandatory for steps 1-3)
+ -c (--config) = config file [CURRENT: "/usr/local/bin/configs/segpy.config.ini"]
+ -V (--verbose) = verbose output
+
+You can import the data and working directory into the container using --bind, and the remaining steps are identical to those outlined in segpy local.
singularity exec --bind ~/seg_cont/outfolder ./segr_cont.img launch_segpy.sh -d ~/seg_cont/outfolder/run --steps 0
+singularity exec --bind ~/seg_cont/outfolder ./segr_cont.img launch_segpy.sh -d ~/seg_cont/outfolder/run --steps 1 --vcf ~/seg_cont/outfolder/data/VEP_iPSC.vcf
+singularity exec --bind ~/seg_cont/outfolder ./segr_cont.img launch_segpy.sh -d ~/seg_cont/outfolder/run --steps 2 --ped ~/seg_cont/outfolder/data/iPSC_2.ped
+singularity exec --bind ~/seg_cont/outfolder ./segr_cont.img launch_segpy.sh -d ~/seg_cont/outfolder/run --steps 3 --parser general
+This is an initial version, and any contributions or suggestions are welcomed. The pipeline is designed to be user-friendly for segregation analysis. For direct contact, please reach out to The Neuro Bioinformatics Core via [neurobioinfo@mcgill.ca].
+If you encounter any issue, please report them on the GitHub repository.
+ +Segpy is a comprehensive pipeline designed for segregation analysis. This documentation provides a step-by-step tutorial on executing segregation analysis in a High-Performance Computing (HPC) environment utilizing the Slurm workload manager system (https://slurm.schedmd.com/), on a Linux workstation, or directly through the segpy module in Python. Segregation analysis is a crucial process for exploring genetic variants within a sample of sequence data. This pipeline facilitates the counting of affected and non-affected individuals with variants, including homozygous variants, those with no variants, and those with no calls. These counts are computed both within families and globally. Additionally, the pipeline offers a detailed breakdown not only of variants but also of alleles in each case. To execute segregation analysis successfully, it is imperative to have a pedigree file with six columns: familyid, individualid, parentalid, maternalid, sex{1:male; 2:female, 0:unknown}, and phenotype={1: control (unaffected), 2: proband(affected), -9:missing}. The genetic data must be provided in the vcf format.
For guidance on how to use segpy's pipeline, consult the tutorial.
+I am HERE.
+segpy is a Python module specifically designed for segregation analysis, developed on Python 3.10.2. The module can be easily downloaded using pip package manager:
pip install 'git+https://github.com/neurobioinfo/segpy#subdirectory=segpy'
+Segregation analysis can be conducted utilizing the segpy scheduler, segpy.pip. If you have access to an HPC or Linux workstation, you can automate the process using segpy.pip. The scheduler script is written in Bash, making it compatible with systems such as Slurm or a Linux workstation.
wget https://github.com/neurobioinfo/segpy/releases/download/v0.2.2.3/segpy.pip.zip
+unzip segpy.pip.zip
+To obtain a brief overview of the pipeline, run the following code.
+bash ./segpy.pip/launch_segpy.sh -h
+The configs/ directory contains the segpy.config.ini file which allows users to specify the parameters of interest explained below
+ACCOUNT Specify user name for slurm module.
+SPARK_PATH Environment variables to point to the correct Spark executable
+ENV_PATH Environment variables to point to the correct Python executable
+PYTHON_CMD=python3.10
+MODULEUSE The path to the environmental module
+JAVA_CMD=java
+JAVA_VERSION=11.0.2
Specify the version of Java
+NCOL=7 To control usage of memory to process CSQ.
+CSQ=True To process CSQ in the output, Consequence annotations from Ensembl VEP.
+GRCH=GRCh38 Genome Reference Consortium Human
+SPARKMEM="16g" Spark Memory
+JAVATOOLOPTIONS="-Xmx8g" Java Memory and any other options can be added to here.
WALLTIME_ARRAY["step_1"]=00-01:00 Set a limit on the total run time of the job allocation in the step 1 under slurm.
+THREADS_ARRAY["step_1"]=4 Request the maximum ntasks be invoked on each core in the step 1 under slurm.
+MEM_ARRAY["step_1"]=25g Specify the real memory required per node in the step 1 under slurm.
WALLTIME_ARRAY["step_2"]=00-02:00 Set a limit on the total run time of the job allocation in the step 2 under slurm.
+THREADS_ARRAY["step_2"]=4 Request the maximum ntasks be invoked on each core in the step 2 under slurm.
+MEM_ARRAY["step_2"]=17g Specify the real memory required per node in the step 2 under slurm.
+INFO_REQUIRED=[1,2,3,4] One can determine the desired content to be included in the output
+ 1: Global affected, 2: Global unaffected, 3: family-wise, 4: phenotype-family-wise, 5: phenotype-family-wise and non-include, 6: phenotype-family-wise-multipe sample, 7: phenotype-family-wise-multipe sample and non-include
WALLTIME_ARRAY["step_3"]=00-01:00 Set a limit on the total run time of the job allocation in the step 3 under slurm.
+THREADS_ARRAY["step_3"]=4 Request the maximum ntasks be invoked on each core in the step 3 under slurm.
+MEM_ARRAY["step_3"]=4g Specify the real memory required per node in the step 3 under slurm.
' + escapeHtml(summary) +'
' + noResultsText + '
'); + } +} + +function doSearch () { + var query = document.getElementById('mkdocs-search-query').value; + if (query.length > min_search_length) { + if (!window.Worker) { + displayResults(search(query)); + } else { + searchWorker.postMessage({query: query}); + } + } else { + // Clear results for short queries + displayResults([]); + } +} + +function initSearch () { + var search_input = document.getElementById('mkdocs-search-query'); + if (search_input) { + search_input.addEventListener("keyup", doSearch); + } + var term = getSearchTermFromLocation(); + if (term) { + search_input.value = term; + doSearch(); + } +} + +function onWorkerMessage (e) { + if (e.data.allowSearch) { + initSearch(); + } else if (e.data.results) { + var results = e.data.results; + displayResults(results); + } else if (e.data.config) { + min_search_length = e.data.config.min_search_length-1; + } +} + +if (!window.Worker) { + console.log('Web Worker API not supported'); + // load index in main thread + $.getScript(joinUrl(base_url, "search/worker.js")).done(function () { + console.log('Loaded worker'); + init(); + window.postMessage = function (msg) { + onWorkerMessage({data: msg}); + }; + }).fail(function (jqxhr, settings, exception) { + console.error('Could not load worker.js'); + }); +} else { + // Wrap search in a web worker + var searchWorker = new Worker(joinUrl(base_url, "search/worker.js")); + searchWorker.postMessage({init: true}); + searchWorker.onmessage = onWorkerMessage; +} diff --git a/0.0.1/search/search_index.json b/0.0.1/search/search_index.json new file mode 100644 index 0000000..31162a5 --- /dev/null +++ b/0.0.1/search/search_index.json @@ -0,0 +1 @@ +{"config":{"indexing":"full","lang":["en"],"min_search_length":3,"prebuild_index":false,"separator":"[\\s\\-]+"},"docs":[{"location":"","text":"Welcome to segpy's documentation! Segpy is a comprehensive pipeline designed for segregation analysis. This documentation provides a step-by-step tutorial on executing segregation analysis in a High-Performance Computing (HPC) environment utilizing the Slurm workload manager system (https://slurm.schedmd.com/), on a Linux workstation, or directly through the segpy module in Python. Segregation analysis is a crucial process for exploring genetic variants within a sample of sequence data. This pipeline facilitates the counting of affected and non-affected individuals with variants, including homozygous variants, those with no variants, and those with no calls. These counts are computed both within families and globally. Additionally, the pipeline offers a detailed breakdown not only of variants but also of alleles in each case. To execute segregation analysis successfully, it is imperative to have a pedigree file with six columns: familyid , individualid , parentalid , maternalid , sex {1:male; 2:female, 0:unknown}, and phenotype ={1: control (unaffected), 2: proband(affected), -9:missing}. The genetic data must be provided in the vcf format. For guidance on how to use segpy's pipeline, consult the tutorial. Contents Installation Tutorial: segpy module segpy slurm segpy local container FAQ Reference","title":"Home"},{"location":"#welcome-to-segpys-documentation","text":"Segpy is a comprehensive pipeline designed for segregation analysis. This documentation provides a step-by-step tutorial on executing segregation analysis in a High-Performance Computing (HPC) environment utilizing the Slurm workload manager system (https://slurm.schedmd.com/), on a Linux workstation, or directly through the segpy module in Python. Segregation analysis is a crucial process for exploring genetic variants within a sample of sequence data. This pipeline facilitates the counting of affected and non-affected individuals with variants, including homozygous variants, those with no variants, and those with no calls. These counts are computed both within families and globally. Additionally, the pipeline offers a detailed breakdown not only of variants but also of alleles in each case. To execute segregation analysis successfully, it is imperative to have a pedigree file with six columns: familyid , individualid , parentalid , maternalid , sex {1:male; 2:female, 0:unknown}, and phenotype ={1: control (unaffected), 2: proband(affected), -9:missing}. The genetic data must be provided in the vcf format. For guidance on how to use segpy's pipeline, consult the tutorial.","title":"Welcome to segpy's documentation!"},{"location":"#contents","text":"Installation Tutorial: segpy module segpy slurm segpy local container FAQ Reference","title":"Contents"},{"location":"Acknowledgement/","text":"Acknowledgement The pipeline is done as part of MNI projects, it is written by Saeid Amiri with associate with Dan Spiegelman, and Sali Farhan at Neuro Bioinformatics Core. Copyright belongs MNI BIOINFO CORE .","title":"- Acknowledgement"},{"location":"Acknowledgement/#acknowledgement","text":"The pipeline is done as part of MNI projects, it is written by Saeid Amiri with associate with Dan Spiegelman, and Sali Farhan at Neuro Bioinformatics Core. Copyright belongs MNI BIOINFO CORE .","title":"Acknowledgement"},{"location":"FAQ/","text":"Frequently asked questions What is finalseg.csv What is finalseg.csv The output of finalseg.csv can be categorized to 1) locus and alleles, 2) CSQ, 3) Global- Non-Affected 4) Global-Affected, 5) Family, 6) Family-Affected 7) Family - Non-affected. If you do not want to have CSQ in the output file, choose CSQ=False . locus and alleles locus: chromosome alleles: a variant form of a gene CSQ VEP put all the requested information in infront CSQ. Global - Non-Affected- is dropt glb_naf_wild: Global - Non-Affecteds, wildtype glb_naf_ncl: Global - Non-Affecteds, no call glb_naf_vrt: Global - Non-Affecteds, with variant glb_naf_homv: Global - Non-Affecteds, homozygous for ALT allele glb_naf_altaf: Global - Non-Affecteds, ALT allele frequency Global - Affected - is dropt glb_aff_wild: Global - Affecteds, wildtype glb_aff_ncl: Global - Affecteds, no call glb_aff_vrt: Global - Affecteds, with variant glb_aff_homv: Global - Affecteds, homozygous for ALT allele glb_aff_altaf: Global - Affecteds, ALT allele frequency Family- Is dropt {famid}_wild: Family - Affecteds: wildtype {famid}_ncl: Family - Affecteds: no call {famid}_vrt: Family - Affecteds: with variant {famid}_homv: Family - Affecteds: homozygous for ALT allele Family - Affected {famid}_aff_wild: Family - Affecteds: wildtype {famid}_aff_ncl: Family - Affecteds: no call {famid}_aff_vrt: Family - Affecteds: with variant {famid}_aff_homv: Family - Affecteds: homozygous for ALT allele Family - Nonaffected {famid}_wild_naf: Family - Nonaffecteds: wildtype {famid}_ncl_naf: Family - Nonaffecteds: no call {famid}_vrt_naf: Family - Nonaffecteds: with variant {famid}_homv_naf: Family - Nonaffecteds: homozygous for ALT allele","title":"FAQ"},{"location":"FAQ/#frequently-asked-questions","text":"What is finalseg.csv","title":"Frequently asked questions"},{"location":"FAQ/#what-is-finalsegcsv","text":"The output of finalseg.csv can be categorized to 1) locus and alleles, 2) CSQ, 3) Global- Non-Affected 4) Global-Affected, 5) Family, 6) Family-Affected 7) Family - Non-affected. If you do not want to have CSQ in the output file, choose CSQ=False .","title":"What is finalseg.csv"},{"location":"FAQ/#locus-and-alleles","text":"locus: chromosome alleles: a variant form of a gene","title":"locus and alleles"},{"location":"FAQ/#csq","text":"VEP put all the requested information in infront CSQ.","title":"CSQ"},{"location":"FAQ/#global-non-affected-is-dropt","text":"glb_naf_wild: Global - Non-Affecteds, wildtype glb_naf_ncl: Global - Non-Affecteds, no call glb_naf_vrt: Global - Non-Affecteds, with variant glb_naf_homv: Global - Non-Affecteds, homozygous for ALT allele glb_naf_altaf: Global - Non-Affecteds, ALT allele frequency","title":"Global - Non-Affected- is dropt"},{"location":"FAQ/#global-affected-is-dropt","text":"glb_aff_wild: Global - Affecteds, wildtype glb_aff_ncl: Global - Affecteds, no call glb_aff_vrt: Global - Affecteds, with variant glb_aff_homv: Global - Affecteds, homozygous for ALT allele glb_aff_altaf: Global - Affecteds, ALT allele frequency","title":"Global - Affected - is dropt"},{"location":"FAQ/#family-is-dropt","text":"{famid}_wild: Family - Affecteds: wildtype {famid}_ncl: Family - Affecteds: no call {famid}_vrt: Family - Affecteds: with variant {famid}_homv: Family - Affecteds: homozygous for ALT allele","title":"Family- Is dropt"},{"location":"FAQ/#family-affected","text":"{famid}_aff_wild: Family - Affecteds: wildtype {famid}_aff_ncl: Family - Affecteds: no call {famid}_aff_vrt: Family - Affecteds: with variant {famid}_aff_homv: Family - Affecteds: homozygous for ALT allele","title":"Family - Affected"},{"location":"FAQ/#family-nonaffected","text":"{famid}_wild_naf: Family - Nonaffecteds: wildtype {famid}_ncl_naf: Family - Nonaffecteds: no call {famid}_vrt_naf: Family - Nonaffecteds: with variant {famid}_homv_naf: Family - Nonaffecteds: homozygous for ALT allele","title":"Family - Nonaffected"},{"location":"LICENSE/","text":"MIT License Copyright (c) 2022 The Neuro Bioinformatics Core Permission is hereby granted, free of charge, to any person obtaining a copy of this software and associated documentation files (the \"Software\"), to deal in the Software without restriction, including without limitation the rights to use, copy, modify, merge, publish, distribute, sublicense, and/or sell copies of the Software, and to permit persons to whom the Software is furnished to do so, subject to the following conditions: The above copyright notice and this permission notice shall be included in all copies or substantial portions of the Software. THE SOFTWARE IS PROVIDED \"AS IS\", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.","title":"- License"},{"location":"about/","text":"Coming soon","title":"Coming soon"},{"location":"about/#coming-soon","text":"","title":"Coming soon"},{"location":"changelog/","text":"Changelog v0.0.1 This is the initial release. v0.0.2 Supposed to Complete the documentation","title":"- Changelog"},{"location":"changelog/#changelog","text":"","title":"Changelog"},{"location":"changelog/#v001","text":"This is the initial release.","title":"v0.0.1"},{"location":"changelog/#v002","text":"Supposed to Complete the documentation","title":"v0.0.2"},{"location":"container/","text":"Container (Under review) The pipeline can be executed using a Singularity container, eliminating the need to install modules within the pipeline. It is compatible for use on High-Performance Computing (HPC) systems or any standard workstation linux system. Contents Step 1: Download Step 2: Run Download Download the segregation image. singularity pull library://saeidamiri1/mni/seg_cont.sif:latest Run Execute singularity exec ./seg_cont.sif launch_segpy.sh -h to ensure the pipeline is functioning correctly. ------------------------------------ Segregation pipline version 0.2.2.3 Usage: /usr/local/bin/launch_segpy.sh [arguments] mandatory arguments: -d (--dir) = Working directory (where all the outputs will be printed) (give full path) -s (--steps) = Specify what steps, e.g., 2 to run just step 2, 1-3 (run steps 1 through 3). 'ALL' to run all steps. steps: 0: initial setup 1: create hail matrix 2: run segregation 3: final cleanup and formatting optional arguments: -h (--help) = Get the program options and exit. --parser = 'general': to general parsing, 'unique': drop multiplicities -v (--vcf) = VCF file (mandatory for steps 1-3) -p (--ped) = PED file (mandatory for steps 1-3) -c (--config) = config file [CURRENT: \"/usr/local/bin/configs/segpy.config.ini\"] -V (--verbose) = verbose output You can import the data and working directory into the container using --bind , and the remaining steps are identical to those outlined in segpy local . singularity exec --bind ~/seg_cont/outfolder ./segr_cont.img launch_segpy.sh -d ~/seg_cont/outfolder/run --steps 0 singularity exec --bind ~/seg_cont/outfolder ./segr_cont.img launch_segpy.sh -d ~/seg_cont/outfolder/run --steps 1 --vcf ~/seg_cont/outfolder/data/VEP_iPSC.vcf singularity exec --bind ~/seg_cont/outfolder ./segr_cont.img launch_segpy.sh -d ~/seg_cont/outfolder/run --steps 2 --ped ~/seg_cont/outfolder/data/iPSC_2.ped singularity exec --bind ~/seg_cont/outfolder ./segr_cont.img launch_segpy.sh -d ~/seg_cont/outfolder/run --steps 3 --parser general \u2b06 back to top","title":"- - container"},{"location":"container/#container-under-review","text":"The pipeline can be executed using a Singularity container, eliminating the need to install modules within the pipeline. It is compatible for use on High-Performance Computing (HPC) systems or any standard workstation linux system.","title":"Container (Under review)"},{"location":"container/#contents","text":"Step 1: Download Step 2: Run","title":"Contents"},{"location":"container/#download","text":"Download the segregation image. singularity pull library://saeidamiri1/mni/seg_cont.sif:latest","title":"Download"},{"location":"container/#run","text":"Execute singularity exec ./seg_cont.sif launch_segpy.sh -h to ensure the pipeline is functioning correctly. ------------------------------------ Segregation pipline version 0.2.2.3 Usage: /usr/local/bin/launch_segpy.sh [arguments] mandatory arguments: -d (--dir) = Working directory (where all the outputs will be printed) (give full path) -s (--steps) = Specify what steps, e.g., 2 to run just step 2, 1-3 (run steps 1 through 3). 'ALL' to run all steps. steps: 0: initial setup 1: create hail matrix 2: run segregation 3: final cleanup and formatting optional arguments: -h (--help) = Get the program options and exit. --parser = 'general': to general parsing, 'unique': drop multiplicities -v (--vcf) = VCF file (mandatory for steps 1-3) -p (--ped) = PED file (mandatory for steps 1-3) -c (--config) = config file [CURRENT: \"/usr/local/bin/configs/segpy.config.ini\"] -V (--verbose) = verbose output You can import the data and working directory into the container using --bind , and the remaining steps are identical to those outlined in segpy local . singularity exec --bind ~/seg_cont/outfolder ./segr_cont.img launch_segpy.sh -d ~/seg_cont/outfolder/run --steps 0 singularity exec --bind ~/seg_cont/outfolder ./segr_cont.img launch_segpy.sh -d ~/seg_cont/outfolder/run --steps 1 --vcf ~/seg_cont/outfolder/data/VEP_iPSC.vcf singularity exec --bind ~/seg_cont/outfolder ./segr_cont.img launch_segpy.sh -d ~/seg_cont/outfolder/run --steps 2 --ped ~/seg_cont/outfolder/data/iPSC_2.ped singularity exec --bind ~/seg_cont/outfolder ./segr_cont.img launch_segpy.sh -d ~/seg_cont/outfolder/run --steps 3 --parser general \u2b06 back to top","title":"Run"},{"location":"contributing/","text":"Contributing This is an initial version, and any contributions or suggestions are welcomed. The pipeline is designed to be user-friendly for segregation analysis. For direct contact, please reach out to The Neuro Bioinformatics Core via [neurobioinfo@mcgill.ca]. If you encounter any issue , please report them on the GitHub repository.","title":"- Contributing"},{"location":"contributing/#contributing","text":"This is an initial version, and any contributions or suggestions are welcomed. The pipeline is designed to be user-friendly for segregation analysis. For direct contact, please reach out to The Neuro Bioinformatics Core via [neurobioinfo@mcgill.ca]. If you encounter any issue , please report them on the GitHub repository.","title":"Contributing"},{"location":"installation/","text":"Installation I am HERE. segpy is a Python module specifically designed for segregation analysis, developed on Python 3.10.2. The module can be easily downloaded using pip package manager: pip install 'git+https://github.com/neurobioinfo/segpy#subdirectory=segpy' Segregation analysis can be conducted utilizing the segpy scheduler, segpy.pip . If you have access to an HPC or Linux workstation, you can automate the process using segpy.pip . The scheduler script is written in Bash, making it compatible with systems such as Slurm or a Linux workstation. wget https://github.com/neurobioinfo/segpy/releases/download/v0.2.2.3/segpy.pip.zip unzip segpy.pip.zip To obtain a brief overview of the pipeline, run the following code. bash ./segpy.pip/launch_segpy.sh -h","title":"Installation"},{"location":"installation/#installation","text":"I am HERE. segpy is a Python module specifically designed for segregation analysis, developed on Python 3.10.2. The module can be easily downloaded using pip package manager: pip install 'git+https://github.com/neurobioinfo/segpy#subdirectory=segpy' Segregation analysis can be conducted utilizing the segpy scheduler, segpy.pip . If you have access to an HPC or Linux workstation, you can automate the process using segpy.pip . The scheduler script is written in Bash, making it compatible with systems such as Slurm or a Linux workstation. wget https://github.com/neurobioinfo/segpy/releases/download/v0.2.2.3/segpy.pip.zip unzip segpy.pip.zip To obtain a brief overview of the pipeline, run the following code. bash ./segpy.pip/launch_segpy.sh -h","title":"Installation"},{"location":"reference/","text":"Adjustable execution parameters for the segregation pipeline Introduction Segpy parameters Introduction The configs/ directory contains the segpy.config.ini file which allows users to specify the parameters of interest explained below Segpy parameters ACCOUNT Specify user name for slurm module. SPARK_PATH Environment variables to point to the correct Spark executable ENV_PATH Environment variables to point to the correct Python executable PYTHON_CMD=python3.10 MODULEUSE The path to the environmental module JAVA_CMD=java JAVA_VERSION=11.0.2 Specify the version of Java NCOL=7 To control usage of memory to process CSQ. CSQ=True To process CSQ in the output, Consequence annotations from Ensembl VEP. GRCH=GRCh38 Genome Reference Consortium Human SPARKMEM=\"16g\" Spark Memory JAVATOOLOPTIONS=\"-Xmx8g\" Java Memory and any other options can be added to here. [step 1] Create table matrix WALLTIME_ARRAY[\"step_1\"]=00-01:00 Set a limit on the total run time of the job allocation in the step 1 under slurm. THREADS_ARRAY[\"step_1\"]=4 Request the maximum ntasks be invoked on each core in the step 1 under slurm. MEM_ARRAY[\"step_1\"]=25g Specify the real memory required per node in the step 1 under slurm. [step 2] Run segregation WALLTIME_ARRAY[\"step_2\"]=00-02:00 Set a limit on the total run time of the job allocation in the step 2 under slurm. THREADS_ARRAY[\"step_2\"]=4 Request the maximum ntasks be invoked on each core in the step 2 under slurm. MEM_ARRAY[\"step_2\"]=17g Specify the real memory required per node in the step 2 under slurm. INFO_REQUIRED=[1,2,3,4] One can determine the desired content to be included in the output 1: Global affected, 2: Global unaffected, 3: family-wise, 4: phenotype-family-wise, 5: phenotype-family-wise and non-include, 6: phenotype-family-wise-multipe sample, 7: phenotype-family-wise-multipe sample and non-include [step 3] Parsing WALLTIME_ARRAY[\"step_3\"]=00-01:00 Set a limit on the total run time of the job allocation in the step 3 under slurm. THREADS_ARRAY[\"step_3\"]=4 Request the maximum ntasks be invoked on each core in the step 3 under slurm. MEM_ARRAY[\"step_3\"]=4g Specify the real memory required per node in the step 3 under slurm.","title":"Reference"},{"location":"reference/#adjustable-execution-parameters-for-the-segregation-pipeline","text":"Introduction Segpy parameters","title":"Adjustable execution parameters for the segregation pipeline"},{"location":"reference/#introduction","text":"The configs/ directory contains the segpy.config.ini file which allows users to specify the parameters of interest explained below","title":"Introduction"},{"location":"reference/#segpy-parameters","text":"ACCOUNT Specify user name for slurm module. SPARK_PATH Environment variables to point to the correct Spark executable ENV_PATH Environment variables to point to the correct Python executable PYTHON_CMD=python3.10 MODULEUSE The path to the environmental module JAVA_CMD=java JAVA_VERSION=11.0.2 Specify the version of Java NCOL=7 To control usage of memory to process CSQ. CSQ=True To process CSQ in the output, Consequence annotations from Ensembl VEP. GRCH=GRCh38 Genome Reference Consortium Human SPARKMEM=\"16g\" Spark Memory JAVATOOLOPTIONS=\"-Xmx8g\" Java Memory and any other options can be added to here.","title":"Segpy parameters"},{"location":"reference/#step-1-create-table-matrix","text":"WALLTIME_ARRAY[\"step_1\"]=00-01:00 Set a limit on the total run time of the job allocation in the step 1 under slurm. THREADS_ARRAY[\"step_1\"]=4 Request the maximum ntasks be invoked on each core in the step 1 under slurm. MEM_ARRAY[\"step_1\"]=25g Specify the real memory required per node in the step 1 under slurm.","title":"[step 1] Create table matrix"},{"location":"reference/#step-2-run-segregation","text":"WALLTIME_ARRAY[\"step_2\"]=00-02:00 Set a limit on the total run time of the job allocation in the step 2 under slurm. THREADS_ARRAY[\"step_2\"]=4 Request the maximum ntasks be invoked on each core in the step 2 under slurm. MEM_ARRAY[\"step_2\"]=17g Specify the real memory required per node in the step 2 under slurm. INFO_REQUIRED=[1,2,3,4] One can determine the desired content to be included in the output 1: Global affected, 2: Global unaffected, 3: family-wise, 4: phenotype-family-wise, 5: phenotype-family-wise and non-include, 6: phenotype-family-wise-multipe sample, 7: phenotype-family-wise-multipe sample and non-include","title":"[step 2] Run segregation"},{"location":"reference/#step-3-parsing","text":"WALLTIME_ARRAY[\"step_3\"]=00-01:00 Set a limit on the total run time of the job allocation in the step 3 under slurm. THREADS_ARRAY[\"step_3\"]=4 Request the maximum ntasks be invoked on each core in the step 3 under slurm. MEM_ARRAY[\"step_3\"]=4g Specify the real memory required per node in the step 3 under slurm.","title":"[step 3] Parsing"},{"location":"segpy_local/","text":"Segpy on workstation segpy.pip is a sheild designed for executing the segpy analysis on Linux workstation or HPC system utilizing the Slurm scheduler. Below, we illustrate the code to submit jobs for (steps 0 to 3) of the pipeline on Linux workstation. Contents Step 0: Setup Step 1: Create table matrix Step 2: Run segregation Step 3: Clean final data The following flowchart illustrates the steps for running the segregation analysis on HPC system utilizing the Slurm system. segpy.svn workflow To execute the pipeline, you require 1) the path of the pipeline (PIPELINE_HOME), 2) the working directory, 3) the VCF file, and 4) the PED file. export PIPELINE_HOME=~/segpy.slurm PWD=~/outfolder VCF=~/data/VEP_iPSC.vcf PED=~/data/iPSC_2.ped Step 0: Setup Initially, execute the following code to set up the pipeline. You can modify the parameters in ${PWD}/job_output/segpy.config.ini. sh $PIPELINE_HOME/launch_pipeline.segpy.sh \\ -d ${PWD} \\ --steps 0 \\ --mode local Step 1: Create table matrix The following code, create MatrixTable from the VCF file. sh $PIPELINE_HOME/launch_pipeline.segpy.sh \\ -d ${PWD} \\ --steps 1 \\ --vcf ${VCF} Step 2: Run segregation Execute the following code to generate the segregation. sh $PIPELINE_HOME/launch_pipeline.segpy.sh \\ -d ${PWD} \\ --steps 2 \\ --vcf ${VCF} \\ --ped ${PED} Step 3: Parsing To parse the file and remove unnecessary characters such as \", [, ], etc., run the following code. sh $PIPELINE_HOME/launch_pipeline.segpy.sh \\ -d ${PWD} \\ --steps 3 \\ --parser general The following code eliminates duplicate information in CSQ. sh $PIPELINE_HOME/launch_pipeline.segpy.sh \\ -d ${PWD} \\ --steps 3 \\ --parser unique Note You can execute steps 1 to 3 sequentially, as illustrated below. sh $PIPELINE_HOME/launch_pipeline.segpy.sh \\ -d ${PWD} \\ --steps 1-3 \\ --vcf ${VCF} \\ --ped ${PED} \\ --parser general \u2b06 back to top","title":"- - segpy local"},{"location":"segpy_local/#segpy-on-workstation","text":"segpy.pip is a sheild designed for executing the segpy analysis on Linux workstation or HPC system utilizing the Slurm scheduler. Below, we illustrate the code to submit jobs for (steps 0 to 3) of the pipeline on Linux workstation.","title":"Segpy on workstation"},{"location":"segpy_local/#contents","text":"Step 0: Setup Step 1: Create table matrix Step 2: Run segregation Step 3: Clean final data The following flowchart illustrates the steps for running the segregation analysis on HPC system utilizing the Slurm system. segpy.svn workflow To execute the pipeline, you require 1) the path of the pipeline (PIPELINE_HOME), 2) the working directory, 3) the VCF file, and 4) the PED file. export PIPELINE_HOME=~/segpy.slurm PWD=~/outfolder VCF=~/data/VEP_iPSC.vcf PED=~/data/iPSC_2.ped","title":"Contents"},{"location":"segpy_local/#step-0-setup","text":"Initially, execute the following code to set up the pipeline. You can modify the parameters in ${PWD}/job_output/segpy.config.ini. sh $PIPELINE_HOME/launch_pipeline.segpy.sh \\ -d ${PWD} \\ --steps 0 \\ --mode local","title":"Step 0: Setup"},{"location":"segpy_local/#step-1-create-table-matrix","text":"The following code, create MatrixTable from the VCF file. sh $PIPELINE_HOME/launch_pipeline.segpy.sh \\ -d ${PWD} \\ --steps 1 \\ --vcf ${VCF}","title":"Step 1: Create table matrix"},{"location":"segpy_local/#step-2-run-segregation","text":"Execute the following code to generate the segregation. sh $PIPELINE_HOME/launch_pipeline.segpy.sh \\ -d ${PWD} \\ --steps 2 \\ --vcf ${VCF} \\ --ped ${PED}","title":"Step 2: Run segregation"},{"location":"segpy_local/#step-3-parsing","text":"To parse the file and remove unnecessary characters such as \", [, ], etc., run the following code. sh $PIPELINE_HOME/launch_pipeline.segpy.sh \\ -d ${PWD} \\ --steps 3 \\ --parser general The following code eliminates duplicate information in CSQ. sh $PIPELINE_HOME/launch_pipeline.segpy.sh \\ -d ${PWD} \\ --steps 3 \\ --parser unique","title":"Step 3: Parsing"},{"location":"segpy_local/#note","text":"You can execute steps 1 to 3 sequentially, as illustrated below. sh $PIPELINE_HOME/launch_pipeline.segpy.sh \\ -d ${PWD} \\ --steps 1-3 \\ --vcf ${VCF} \\ --ped ${PED} \\ --parser general \u2b06 back to top","title":"Note"},{"location":"segpy_module/","text":"Segpy module The following steps illustrate how to execute the segregation pipeline directly by utilizing the Segpy module within the Python editor. segpy workflow Contents Step 1: Run Spark Step 2: Create table matrix Step 3: Run segregation Step 4: Parsing Step 5: Shut down spark Step 1: Run Spark Initially, activate Spark on your system. export SPARK_HOME=$HOME/spark-3.1.3-bin-hadoop3.2 export SPARK_LOG_DIR=$HOME/temp module load java/11.0.2 ${SPARK_HOME}/sbin/start-master.sh Step 2: Create table matrix Following that, initialize Hail, import your VCF file, and save it as a matrix table. The matrix table serves as a data structure for representing genetic data in matrix form. Below, we import the VCF file, save it as a MatrixTable, and proceed to read your matrix table. In the tutorial, additional data is incorporated to test the pipeline, which can be found at [https://github.com/The-Neuro-Bioinformatics-Core/segpy/data]. This dataset is utilized to illustrate the pipeline's functionality. The subsequent code imports a VCF file and represents it as a MatrixTable: import sys import pandas as pd import hail as hl hl.import_vcf('~/test/data/VEP.iPSC.vcf',force=True,reference_genome='GRCh38',array_elements_required=False).write('~/test/output/VEP.iPSC.mt', overwrite=True) mt = hl.read_matrix_table('~/test/output/VEP.iPSC.mt') Step 3: Run segregation Execute the following code to generate the segregation. from segpy import seg ped=pd.read_csv('~/test/data/iPSC_2.ped'.ped',sep='\\t') destfolder= '~/test/output/' vcffile='~/test/data/VEP.iPSC.vcf' ncol=7 CSQ=True seg.run(mt,ped,outfolder,hl,ncol,CSQ,vcffile) Running the provided code generates two files, namely header.txt and finalseg.csv , in the destfolder . The header.txt file contains the header information found in finalseg.csv . The output in finalseg.csv is organized into the following categories: 1) locus and alleles, 2) CSQ, 3) Global-Non-Affected, 4) Global-Affected, 5) Family, 6) Family-Affected, and 7) Family-Non-Affected. If you prefer not to include CSQ in the output file, you can select CSQ=False . Step 4: Parsing To parse the file and remove unnecessary characters such as \", [, ], etc., run the following code. from segpy import parser parser.clean_general(outfolder) The information consists of various components; to eliminate redundancy, execute the following code. parser.clean_unique(outfolder) Step 5: Shut down spark Do not forget to deactivate environment and stop the spark: ${SPARK_HOME}/sbin/stop-master.sh \u2b06 back to top","title":"- - segpy module"},{"location":"segpy_module/#segpy-module","text":"The following steps illustrate how to execute the segregation pipeline directly by utilizing the Segpy module within the Python editor. segpy workflow","title":"Segpy module"},{"location":"segpy_module/#contents","text":"Step 1: Run Spark Step 2: Create table matrix Step 3: Run segregation Step 4: Parsing Step 5: Shut down spark","title":"Contents"},{"location":"segpy_module/#step-1-run-spark","text":"Initially, activate Spark on your system. export SPARK_HOME=$HOME/spark-3.1.3-bin-hadoop3.2 export SPARK_LOG_DIR=$HOME/temp module load java/11.0.2 ${SPARK_HOME}/sbin/start-master.sh","title":"Step 1: Run Spark"},{"location":"segpy_module/#step-2-create-table-matrix","text":"Following that, initialize Hail, import your VCF file, and save it as a matrix table. The matrix table serves as a data structure for representing genetic data in matrix form. Below, we import the VCF file, save it as a MatrixTable, and proceed to read your matrix table. In the tutorial, additional data is incorporated to test the pipeline, which can be found at [https://github.com/The-Neuro-Bioinformatics-Core/segpy/data]. This dataset is utilized to illustrate the pipeline's functionality. The subsequent code imports a VCF file and represents it as a MatrixTable: import sys import pandas as pd import hail as hl hl.import_vcf('~/test/data/VEP.iPSC.vcf',force=True,reference_genome='GRCh38',array_elements_required=False).write('~/test/output/VEP.iPSC.mt', overwrite=True) mt = hl.read_matrix_table('~/test/output/VEP.iPSC.mt')","title":"Step 2: Create table matrix"},{"location":"segpy_module/#step-3-run-segregation","text":"Execute the following code to generate the segregation. from segpy import seg ped=pd.read_csv('~/test/data/iPSC_2.ped'.ped',sep='\\t') destfolder= '~/test/output/' vcffile='~/test/data/VEP.iPSC.vcf' ncol=7 CSQ=True seg.run(mt,ped,outfolder,hl,ncol,CSQ,vcffile) Running the provided code generates two files, namely header.txt and finalseg.csv , in the destfolder . The header.txt file contains the header information found in finalseg.csv . The output in finalseg.csv is organized into the following categories: 1) locus and alleles, 2) CSQ, 3) Global-Non-Affected, 4) Global-Affected, 5) Family, 6) Family-Affected, and 7) Family-Non-Affected. If you prefer not to include CSQ in the output file, you can select CSQ=False .","title":"Step 3: Run segregation"},{"location":"segpy_module/#step-4-parsing","text":"To parse the file and remove unnecessary characters such as \", [, ], etc., run the following code. from segpy import parser parser.clean_general(outfolder) The information consists of various components; to eliminate redundancy, execute the following code. parser.clean_unique(outfolder)","title":"Step 4: Parsing"},{"location":"segpy_module/#step-5-shut-down-spark","text":"Do not forget to deactivate environment and stop the spark: ${SPARK_HOME}/sbin/stop-master.sh \u2b06 back to top","title":"Step 5: Shut down spark"},{"location":"segpy_slurm/","text":"Segpy via slurm segpy.pip is a sheild designed for executing the segpy analysis on Linux workstation or HPC system utilizing the Slurm scheduler. Below, we illustrate the code to submit jobs for (steps 0 to 3) of the pipeline on an HPC system utilizing the Slurm scheduler. This pipeline has been employed on Beluga , an HPC system utilizing the Slurm system. Contents Step 0: Setup Step 1: Create table matrix Step 2: Run segregation Step 3: Clean final data The following flowchart illustrates the steps for running the segregation analysis on HPC system utilizing the Slurm system. segpy.svn workflow To execute the pipeline, you require 1) the path of the pipeline (PIPELINE_HOME), 2) the working directory, 3) the VCF file, and 4) the PED file. export PIPELINE_HOME=~/segpy.slurm PWD=~/outfolder VCF=~/data/VEP_iPSC.vcf PED=~/data/iPSC_2.ped Step 0: Setup Initially, execute the following code to set up the pipeline. You can modify the parameters in ${PWD}/job_output/segpy.config.ini. sh $PIPELINE_HOME/launch_pipeline.segpy.sh \\ -d ${PWD} \\ --steps 0 \\ --mode slurm Step 1: Create table matrix The following code, create MatrixTable from the VCF file. sh $PIPELINE_HOME/launch_pipeline.segpy.sh \\ -d ${PWD} \\ --steps 1 \\ --vcf ${VCF} Step 2: Run segregation Execute the following code to generate the segregation. sh $PIPELINE_HOME/launch_pipeline.segpy.sh \\ -d ${PWD} \\ --steps 2 \\ --vcf ${VCF} \\ --ped ${PED} Step 3: Parsing To parse the file and remove unnecessary characters such as \", [, ], etc., run the following code. sh $PIPELINE_HOME/launch_pipeline.segpy.sh \\ -d ${PWD} \\ --steps 3 \\ --parser general The following code eliminates duplicate information in CSQ. sh $PIPELINE_HOME/launch_pipeline.segpy.sh \\ -d ${PWD} \\ --steps 3 \\ --parser unique Note You can execute steps 1 to 3 sequentially, as illustrated below. sh $PIPELINE_HOME/launch_pipeline.segpy.sh \\ -d ${PWD} \\ --steps 1-3 \\ --vcf ${VCF} \\ --ped ${PED} \\ --parser general \u2b06 back to top","title":"- - segpy slurm"},{"location":"segpy_slurm/#segpy-via-slurm","text":"segpy.pip is a sheild designed for executing the segpy analysis on Linux workstation or HPC system utilizing the Slurm scheduler. Below, we illustrate the code to submit jobs for (steps 0 to 3) of the pipeline on an HPC system utilizing the Slurm scheduler. This pipeline has been employed on Beluga , an HPC system utilizing the Slurm system.","title":"Segpy via slurm"},{"location":"segpy_slurm/#contents","text":"Step 0: Setup Step 1: Create table matrix Step 2: Run segregation Step 3: Clean final data The following flowchart illustrates the steps for running the segregation analysis on HPC system utilizing the Slurm system. segpy.svn workflow To execute the pipeline, you require 1) the path of the pipeline (PIPELINE_HOME), 2) the working directory, 3) the VCF file, and 4) the PED file. export PIPELINE_HOME=~/segpy.slurm PWD=~/outfolder VCF=~/data/VEP_iPSC.vcf PED=~/data/iPSC_2.ped","title":"Contents"},{"location":"segpy_slurm/#step-0-setup","text":"Initially, execute the following code to set up the pipeline. You can modify the parameters in ${PWD}/job_output/segpy.config.ini. sh $PIPELINE_HOME/launch_pipeline.segpy.sh \\ -d ${PWD} \\ --steps 0 \\ --mode slurm","title":"Step 0: Setup"},{"location":"segpy_slurm/#step-1-create-table-matrix","text":"The following code, create MatrixTable from the VCF file. sh $PIPELINE_HOME/launch_pipeline.segpy.sh \\ -d ${PWD} \\ --steps 1 \\ --vcf ${VCF}","title":"Step 1: Create table matrix"},{"location":"segpy_slurm/#step-2-run-segregation","text":"Execute the following code to generate the segregation. sh $PIPELINE_HOME/launch_pipeline.segpy.sh \\ -d ${PWD} \\ --steps 2 \\ --vcf ${VCF} \\ --ped ${PED}","title":"Step 2: Run segregation"},{"location":"segpy_slurm/#step-3-parsing","text":"To parse the file and remove unnecessary characters such as \", [, ], etc., run the following code. sh $PIPELINE_HOME/launch_pipeline.segpy.sh \\ -d ${PWD} \\ --steps 3 \\ --parser general The following code eliminates duplicate information in CSQ. sh $PIPELINE_HOME/launch_pipeline.segpy.sh \\ -d ${PWD} \\ --steps 3 \\ --parser unique","title":"Step 3: Parsing"},{"location":"segpy_slurm/#note","text":"You can execute steps 1 to 3 sequentially, as illustrated below. sh $PIPELINE_HOME/launch_pipeline.segpy.sh \\ -d ${PWD} \\ --steps 1-3 \\ --vcf ${VCF} \\ --ped ${PED} \\ --parser general \u2b06 back to top","title":"Note"},{"location":"tutorial/","text":"Tutorial This section contains tutorials for segregation analysis. segpy module segpy slurm segpy local container","title":"Tutorial"},{"location":"tutorial/#tutorial","text":"This section contains tutorials for segregation analysis.","title":"Tutorial"},{"location":"tutorial/#segpy-module","text":"","title":"segpy module"},{"location":"tutorial/#segpy-slurm","text":"","title":"segpy slurm"},{"location":"tutorial/#segpy-local","text":"","title":"segpy local"},{"location":"tutorial/#container","text":"","title":"container"}]} \ No newline at end of file diff --git a/0.0.1/search/worker.js b/0.0.1/search/worker.js new file mode 100644 index 0000000..8628dbc --- /dev/null +++ b/0.0.1/search/worker.js @@ -0,0 +1,133 @@ +var base_path = 'function' === typeof importScripts ? '.' : '/search/'; +var allowSearch = false; +var index; +var documents = {}; +var lang = ['en']; +var data; + +function getScript(script, callback) { + console.log('Loading script: ' + script); + $.getScript(base_path + script).done(function () { + callback(); + }).fail(function (jqxhr, settings, exception) { + console.log('Error: ' + exception); + }); +} + +function getScriptsInOrder(scripts, callback) { + if (scripts.length === 0) { + callback(); + return; + } + getScript(scripts[0], function() { + getScriptsInOrder(scripts.slice(1), callback); + }); +} + +function loadScripts(urls, callback) { + if( 'function' === typeof importScripts ) { + importScripts.apply(null, urls); + callback(); + } else { + getScriptsInOrder(urls, callback); + } +} + +function onJSONLoaded () { + data = JSON.parse(this.responseText); + var scriptsToLoad = ['lunr.js']; + if (data.config && data.config.lang && data.config.lang.length) { + lang = data.config.lang; + } + if (lang.length > 1 || lang[0] !== "en") { + scriptsToLoad.push('lunr.stemmer.support.js'); + if (lang.length > 1) { + scriptsToLoad.push('lunr.multi.js'); + } + if (lang.includes("ja") || lang.includes("jp")) { + scriptsToLoad.push('tinyseg.js'); + } + for (var i=0; i < lang.length; i++) { + if (lang[i] != 'en') { + scriptsToLoad.push(['lunr', lang[i], 'js'].join('.')); + } + } + } + loadScripts(scriptsToLoad, onScriptsLoaded); +} + +function onScriptsLoaded () { + console.log('All search scripts loaded, building Lunr index...'); + if (data.config && data.config.separator && data.config.separator.length) { + lunr.tokenizer.separator = new RegExp(data.config.separator); + } + + if (data.index) { + index = lunr.Index.load(data.index); + data.docs.forEach(function (doc) { + documents[doc.location] = doc; + }); + console.log('Lunr pre-built index loaded, search ready'); + } else { + index = lunr(function () { + if (lang.length === 1 && lang[0] !== "en" && lunr[lang[0]]) { + this.use(lunr[lang[0]]); + } else if (lang.length > 1) { + this.use(lunr.multiLanguage.apply(null, lang)); // spread operator not supported in all browsers: https://developer.mozilla.org/en-US/docs/Web/JavaScript/Reference/Operators/Spread_operator#Browser_compatibility + } + this.field('title'); + this.field('text'); + this.ref('location'); + + for (var i=0; i < data.docs.length; i++) { + var doc = data.docs[i]; + this.add(doc); + documents[doc.location] = doc; + } + }); + console.log('Lunr index built, search ready'); + } + allowSearch = true; + postMessage({config: data.config}); + postMessage({allowSearch: allowSearch}); +} + +function init () { + var oReq = new XMLHttpRequest(); + oReq.addEventListener("load", onJSONLoaded); + var index_path = base_path + '/search_index.json'; + if( 'function' === typeof importScripts ){ + index_path = 'search_index.json'; + } + oReq.open("GET", index_path); + oReq.send(); +} + +function search (query) { + if (!allowSearch) { + console.error('Assets for search still loading'); + return; + } + + var resultDocuments = []; + var results = index.search(query); + for (var i=0; i < results.length; i++){ + var result = results[i]; + doc = documents[result.ref]; + doc.summary = doc.text.substring(0, 200); + resultDocuments.push(doc); + } + return resultDocuments; +} + +if( 'function' === typeof importScripts ) { + onmessage = function (e) { + if (e.data.init) { + init(); + } else if (e.data.query) { + postMessage({ results: search(e.data.query) }); + } else { + console.error("Worker - Unrecognized message: " + e); + } + }; +} diff --git a/0.0.1/segpy_local/index.html b/0.0.1/segpy_local/index.html new file mode 100644 index 0000000..93169d6 --- /dev/null +++ b/0.0.1/segpy_local/index.html @@ -0,0 +1,242 @@ + + + + + + + +segpy.pip is a sheild designed for executing the segpy analysis on Linux workstation or HPC system utilizing the Slurm scheduler. Below, we illustrate the code to submit jobs for (steps 0 to 3) of the pipeline on Linux workstation.
The following flowchart illustrates the steps for running the segregation analysis on HPC system utilizing the Slurm system.
+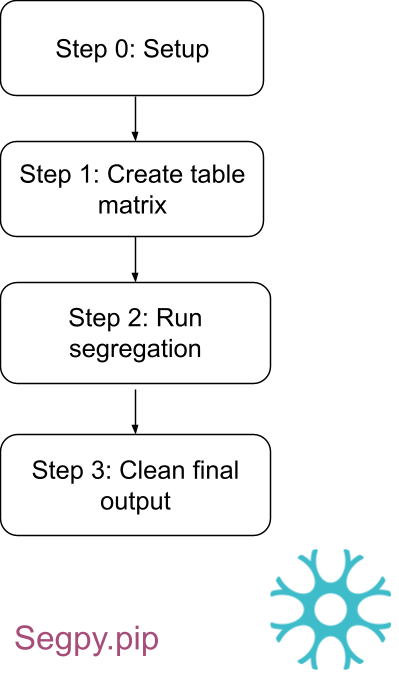 |
+
|---|
| segpy.svn workflow | +
To execute the pipeline, you require 1) the path of the pipeline (PIPELINE_HOME), 2) the working directory, 3) the VCF file, and 4) the PED file.
+export PIPELINE_HOME=~/segpy.slurm
+PWD=~/outfolder
+VCF=~/data/VEP_iPSC.vcf
+PED=~/data/iPSC_2.ped
+Initially, execute the following code to set up the pipeline. You can modify the parameters in ${PWD}/job_output/segpy.config.ini.
+sh $PIPELINE_HOME/launch_pipeline.segpy.sh \
+-d ${PWD} \
+--steps 0 \
+--mode local
+The following code, create MatrixTable from the VCF file.
+sh $PIPELINE_HOME/launch_pipeline.segpy.sh \
+-d ${PWD} \
+--steps 1 \
+--vcf ${VCF}
+Execute the following code to generate the segregation.
+sh $PIPELINE_HOME/launch_pipeline.segpy.sh \
+-d ${PWD} \
+--steps 2 \
+--vcf ${VCF} \
+--ped ${PED}
+To parse the file and remove unnecessary characters such as ", [, ], etc., run the following code.
+sh $PIPELINE_HOME/launch_pipeline.segpy.sh \
+-d ${PWD} \
+--steps 3 \
+--parser general
+The following code eliminates duplicate information in CSQ.
+sh $PIPELINE_HOME/launch_pipeline.segpy.sh \
+-d ${PWD} \
+--steps 3 \
+--parser unique
+You can execute steps 1 to 3 sequentially, as illustrated below.
+sh $PIPELINE_HOME/launch_pipeline.segpy.sh \
+-d ${PWD} \
+--steps 1-3 \
+--vcf ${VCF} \
+--ped ${PED} \
+--parser general
+The following steps illustrate how to execute the segregation pipeline directly by utilizing the Segpy module within the Python editor.
+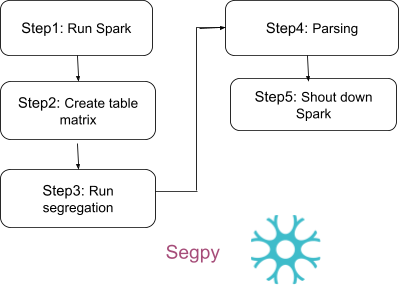 |
+
|---|
| segpy workflow | +
Initially, activate Spark on your system.
+export SPARK_HOME=$HOME/spark-3.1.3-bin-hadoop3.2
+export SPARK_LOG_DIR=$HOME/temp
+module load java/11.0.2
+${SPARK_HOME}/sbin/start-master.sh
+Following that, initialize Hail, import your VCF file, and save it as a matrix table. The matrix table serves as a data structure for representing genetic data in matrix form. Below, we import the VCF file, save it as a MatrixTable, and proceed to read your matrix table. In the tutorial, additional data is incorporated to test the pipeline, which can be found at [https://github.com/The-Neuro-Bioinformatics-Core/segpy/data]. This dataset is utilized to illustrate the pipeline's functionality.
+The subsequent code imports a VCF file and represents it as a MatrixTable:
+import sys
+import pandas as pd
+import hail as hl
+hl.import_vcf('~/test/data/VEP.iPSC.vcf',force=True,reference_genome='GRCh38',array_elements_required=False).write('~/test/output/VEP.iPSC.mt', overwrite=True)
+mt = hl.read_matrix_table('~/test/output/VEP.iPSC.mt')
+Execute the following code to generate the segregation.
+from segpy import seg
+ped=pd.read_csv('~/test/data/iPSC_2.ped'.ped',sep='\t')
+destfolder= '~/test/output/'
+vcffile='~/test/data/VEP.iPSC.vcf'
+ncol=7
+CSQ=True
+seg.run(mt,ped,outfolder,hl,ncol,CSQ,vcffile)
+Running the provided code generates two files, namely header.txt and finalseg.csv, in the destfolder. The header.txt file contains the header information found in finalseg.csv. The output in finalseg.csv is organized into the following categories: 1) locus and alleles, 2) CSQ, 3) Global-Non-Affected, 4) Global-Affected, 5) Family, 6) Family-Affected, and 7) Family-Non-Affected. If you prefer not to include CSQ in the output file, you can select CSQ=False.
To parse the file and remove unnecessary characters such as ", [, ], etc., run the following code.
+from segpy import parser
+parser.clean_general(outfolder)
+The information consists of various components; to eliminate redundancy, execute the following code.
+parser.clean_unique(outfolder)
+Do not forget to deactivate environment and stop the spark:
+${SPARK_HOME}/sbin/stop-master.sh
+segpy.pip is a sheild designed for executing the segpy analysis on Linux workstation or HPC system utilizing the Slurm scheduler. Below, we illustrate the code to submit jobs for (steps 0 to 3) of the pipeline on an HPC system utilizing the Slurm scheduler. This pipeline has been employed on Beluga, an HPC system utilizing the Slurm system.
The following flowchart illustrates the steps for running the segregation analysis on HPC system utilizing the Slurm system.
+ |
+
|---|
| segpy.svn workflow | +
To execute the pipeline, you require 1) the path of the pipeline (PIPELINE_HOME), 2) the working directory, 3) the VCF file, and 4) the PED file.
+export PIPELINE_HOME=~/segpy.slurm
+PWD=~/outfolder
+VCF=~/data/VEP_iPSC.vcf
+PED=~/data/iPSC_2.ped
+Initially, execute the following code to set up the pipeline. You can modify the parameters in ${PWD}/job_output/segpy.config.ini.
+sh $PIPELINE_HOME/launch_pipeline.segpy.sh \
+-d ${PWD} \
+--steps 0 \
+--mode slurm
+The following code, create MatrixTable from the VCF file.
+sh $PIPELINE_HOME/launch_pipeline.segpy.sh \
+-d ${PWD} \
+--steps 1 \
+--vcf ${VCF}
+Execute the following code to generate the segregation.
+sh $PIPELINE_HOME/launch_pipeline.segpy.sh \
+-d ${PWD} \
+--steps 2 \
+--vcf ${VCF} \
+--ped ${PED}
+To parse the file and remove unnecessary characters such as ", [, ], etc., run the following code.
+sh $PIPELINE_HOME/launch_pipeline.segpy.sh \
+-d ${PWD} \
+--steps 3 \
+--parser general
+The following code eliminates duplicate information in CSQ.
+sh $PIPELINE_HOME/launch_pipeline.segpy.sh \
+-d ${PWD} \
+--steps 3 \
+--parser unique
+You can execute steps 1 to 3 sequentially, as illustrated below.
+sh $PIPELINE_HOME/launch_pipeline.segpy.sh \
+-d ${PWD} \
+--steps 1-3 \
+--vcf ${VCF} \
+--ped ${PED} \
+--parser general
+This section contains tutorials for segregation analysis.
+