Home
Currently we are at the experimental release stage, we will be happy if you try it. Please report to Issue Tracker system or write to us in you have problems:
-
Artsemi Yushkevich (Contributing Developer) - Ph.D. student in the In situ Structural Biology Group at the MDC Berlin (Max Delbrück Center of Molecular Medicine) in Berlin, Germany and/or
-
Misha Kudryashev (Project Leader) - Principal Investigator, head of the In situ Structural Biology Group at the MDC Berlin (Max Delbrück Center of Molecular Medicine) in Berlin, Germany.
This wiki contains...
- general description (below on this page)
- description on how to install and setup TomoBEAR
- list of Frequently Asked Questions
- tutorial as the example of TomoBEAR project preparation and execution
- description on all the available modules, their parameters and default values
- TomoBEAR usage tips
TomoBEAR is a configurable and customizable software package specialized for cryo-electron tomography and subtomogram averaging completely written in the MATLAB scripting language. TomoBEAR implements Basics for Electron tomography and Automated Reconstruction of cryo electron tomography data. With TomoBEAR you can easily process tomographic data acquired by an electron microscope starting from the raw tilt series, possibly dose fractionated, or already assembled tilt stacks to the biological structure of interest.
TomoBEAR is designed to operate on a data set in parallel where it is possible and that the user has minimal intervention and doesn't need to learn all the different software packages (MotionCor2, IMOD, Gctf, Dynamo) to be able to process cryo-electron tomography data. We tried to come up with a set of predefined defaults which should fit many projects in the cryo-electron tomography regime. However if some parameters need to be tweaked for specific projects to achieve best results you are free to do so.
List of reasons to use TomoBEAR
- TomoBEAR is based on best practices in processing tomographic data
- Uses default presets which work on a variety of tested datasets
- Standalone and MATLAB versions are available
- Parallel execution is possible
- Computing resources are configurable
- Supports SLURM cluster scheduler
- Standardized folder structure
- Can deal with
- misnumbered tilt images due to SerialEM crashes, based on timestamps
- different naming conventions
- EERs, MRCs and TIFs from K2 and K3
- duplicated projections due to tracking issues (first, last, keep)
- Restarting / resuming is possible (e.g. in case of errors, wrong configuration)
- Checkpoints are created after every processing step of a tilt series or tomogram
- TomoBEAR is based on JSON configuration files which can be easily shared between others so that they can validate or improve your results
- TomoBEAR was developed and tested on standard datasets to achieve same or better results as with manual processing
- You are able to look at the intermediates optimize parameters and rerun the steps to achieve optimal results
- You are never locked to the tomoBEAR processing pipeline and can easily breakout at various steps to other software tools you prefer
- Uses links where possible
- Clean up functionality to save storage
- Tomograms to be processed can be limited to a subset
- Uses Dynamo tilt-series alignment but injects the fiducial positions to IMOD for projection estimation
- Alignment parameters for tilt series can be (optionally) manually refined
- Routines for particle identification: template matching or geometry-assisted particle picking (with Dynamo)
- Improved Dynamo template matching functionality
- 10x - 14x speedup leveraging the GPU compared to 28 CPUs achieving 18x speedup
- Sub-stacking analysis (by SUSAN) is integrated (work in progress)
- TemplateMatchingPostprocessing
- Extraction of particles and conversion to dboxes to speed up file system access speed
- DynamoAlignmentProject
- Classification by multi-reference alignment (MRA)
- Generation of initial templates with with true structures and "noise traps"
- Extraction of particles with SUSAN and conversion to dboxes now possible
- Forcing of using SUSAN is also possible
- BinStacks module with appropriate binning levels should be run upfront
- Modification of particles box size is possible, as input can be used a factor, the absolute box size can be used as input
- Classification by multi-reference alignment (MRA)
- Introduced presorting for single numbered tilt series data (as in Wenbo’s case of published Ryanodine receptor data)
- Automated exclusion of bad tilts in reconstructions based on refined fiducial file from BatchRunTomo module
In the following picture you can see a flow chart which visualizes which steps TomoBEAR will and can do for you in an automated and parallel manner. TomoBEAR supports workstations and single interactive nodes with GPUs on the comuting clusters at the moment. We are also working towards enabling the support of computer clusters through a queue manager like SLURM or SGE (Sun Grid Engine).
Note that it is not needed to start from raw data but you can also use already assembled tilt stacks if you provide the angular information.


Orange processing steps in the flow chart are mandatory and must be executed by TomoBEAR. Yellow boxes are optional and can be activated if desired.
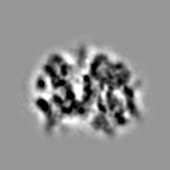
The result shown here have been achieved in an automated manner. The only manual task which needed to be done is the choice of the classes for further processing in between the transition of different binning levels.
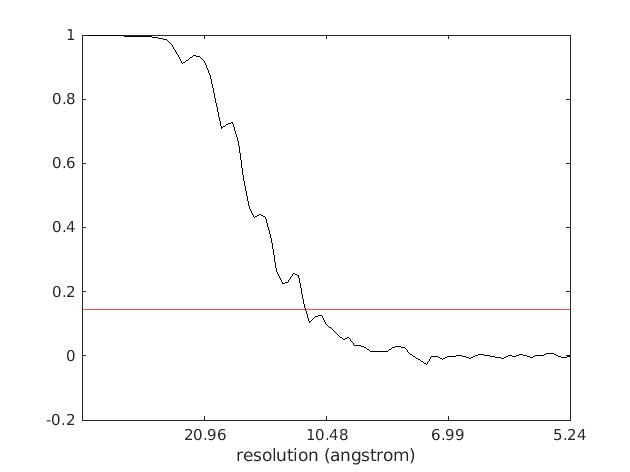
On the EMPIAR-10064 dataset TomoBEAR achieved 11.25 Angstrom (with ~4k particles) as can be seen below on the FSC curve plot:
As well, below is provided ribosome final map view: