Tutorials
This page contains a range of tutorials explaining installation, setup, usage and example output to help you get started with TomoBEAR.
- Setup TomoBEAR
- Tutorial I. 80S ribosomes (EMPIAR-10064)
- Tutorial II. Ryanodine Receptor Type 1 (EMPIAR-10452)
- Tutorial III. In situ 80S ribosomes (EMPIAR-11306)
First of all, before proceeding with tutorials, you have to setup TomoBEAR:
-
Video-tutorial explaining how to get the latest
TomoBEARversion and configureTomoBEARand its dependencies.
In the obtained TomoBEAR folder you may find folder tutorials folder, which contains download/configuration files and some other materials needed for the tutorials, provided on this page:
-
empiar10064_ribo- materials for Tutorial I. 80S ribosomes (EMPIAR-10064) -
empiar10452_ryr1- materials for Tutorial II. Ryanodine Receptor Type 1 (EMPIAR-10452) -
empiar11306_ribo_pfib- materials for Tutorial III. In situ 80S ribosomes (EMPIAR-11306)
To provide an example of the TomoBEAR processing project we have chosen the well-known cryo-ET benchmarking data set containing 80S ribosomes - EMPIAR-10064.
In this tutorial we will:
- explain structure and important parameters of the input
JSONfile used as a configuration file inTomoBEARprojects; - explain output data structure of
TomoBEARprojects; - explain pipeline execution and control by checkpoint files;
- guide you through the processing of the famous EMPIAR-10064 dataset to achieve resolution of 11.3 Å with ~4k particles in nearly-automated parallel manner.
The tutorial materials folder TomoBEAR/tutorials/empiar10064_ribo/ contains:
-
empiar10064_download.sh- data download bash script; -
empiar10064_config.json- processing project JSON configuration file.
We have prepared a range of short (8-12 min) video-tutorials for the 80S ribosome tutorial provided below:
- Video-tutoral 1: description of the project configuration file and the pipeline execution
-
Video-tutoral 2: additional configuration file parameters description,
TomoBEAR-IMOD-TomoBEARloop for checking tilt series alignment results and fiducials refinement (if needed); - Video-tutoral 3: checking on further intermediate results (alignment, CTF-estimation, reconstruction, template matching).
The EMPIAR-10064 data set is located here: https://www.ebi.ac.uk/empiar/EMPIAR-10064. For this tutorial we will need just the mixedCTEM data part.
Note
ThemixedCTEMdata is sufficient to be able to pick ~4k particles achieving resolution of 11.3 Å, which is comparable to the 11.0 Å obtained with the same dataset in the original study (Khoshouei M. et al. (2017) J Struct Biol).
In order to download data you can:
- use EMPIAR interface to select
mixedCTEMdata (4 tilt stacks in mrc file format) and Download button to obtain the corresponding archived data. - use the suggested download script empiar10064_download.sh (located at
tutorials/empiar_10064_ribo):
DATA_DIR=$(readlink -f ./empiar10064.raw)
mkdir ${DATA_DIR}
wget -nH --cut-dirs=4 --progress=bar:force:noscroll -m -P ${DATA_DIR} ftp://ftp.ebi.ac.uk/empiar/world_availability/10064/data/mixedCTEM*.mrcTo use the script, make it executable by
chmod +x ./empiar10064_download.shand run it as
./empiar10064_download.shThe script above should download needed for this tutorial mixedCTEM part of EMPIAR-10064 data set in empiar10064.raw directory.
In order to setup TomoBEAR to process the wished data, the JSON configuration file, describing the processing pipeline and the used parameters, should be prepared. For this tutorial the corresponding configuration file empiar10064_config.json is provided at tutorials/empiar_10064_ribo.
The following paragraphs will explain the structure, components and variables contained in the JSON file and the needed changes to be able to run TomoBEAR on your local machine with this particular dataset.
First of all, you need to setup the following two paths:
-
"data_path"- path to the (raw) input data (which should point toempiar10064.raw/folder if you used our download script); -
"processing_path"- path to the project output folder location (whereTomoBEARproject output folder calledoutput/will be created)
in the section "general": {} of the JSON file, presented below:
"general": {
"project_name": "Ribosome",
"project_description": "Ribosome EMPIAR 10064",
"data_path": "/path/to/raw/data/empiar10064.raw/*.mrc",
"processing_path": "/path/to/your/processing/folder",
"expected_symmetrie": "C1",
"apix": 2.62,
"tilt_angles": [-60.0, -58.0, -56.0, -54.0, -52.0, -50.0, -48.0, -46.0, -44.0, -42.0, -40.0, -38.0, -36.0, -34.0, -32.0, -30.0, -28.0, -26.0, -24.0, -22.0, -20.0, -18.0, -16.0, -14.0, -12.0, -10.0, -8.0, -6.0, -4.0, -2.0, 0.0, 2.0, 4.0, 6.0, 8.0, 10.0, 12.0, 14.0, 16.0, 18.0, 20.0, 22.0, 24.0, 26.0, 28.0, 30.0, 32.0, 34.0, 36.0, 38.0, 40.0, 42.0, 44.0, 46.0, 48.0, 50.0, 52.0, 54.0, 56.0],
"rotation_tilt_axis":-5,
"gold_bead_size_in_nm": 9,
"template_matching_binning": 8,
"binnings": [2, 4, 8],
"reconstruction_thickness": 800,
"gpu": [x,x,x,x],
"as_boxes": false
}The input data we use in this tutorial are four motion-corrected assembled tilt stacks. However, since the pixel size ("apix") and tilt angles ("tilt_angles") are not provided in the header of the corresponding input MRC files, we have to add this information in the "general": {} section of the configuration file as well.
As well, at this stage we also should setup (known) value for expected gold fiducials size ("gold_bead_size_in_nm").
Next, we need to roughly estimate in-plane (XY) tilt-axis rotation and set the value for the corresponding parameter ("rotation_tilt_axis"). It should be sufficient just to set either 80 or -5 degrees, according to initial tilt axis orientation (horizontal or vertical), since this value would be further automatically refined.
Finally, we need to setup available/wished GPU ID's to be used in the corresponding parameter ("gpu"). Note that in TomoBEAR you need to put GPU indexes starting from 1 (so, +1 to GPU indexing from nvidia-smi command output).
The other parameters will be explained later.
At this point the TomoBEAR execution can be started. To run the TomoBEAR on the ribosome data set you need to type in the following command in the MATLAB command window
runTomoBear("local", "/path/to/empiar10064_config.json")The TomoBEAR should run up to the first appearance of StopPipeline block, which will stop further pipeline execution for one time. That means only the following modules will be automatically executed:
"MetaData": {
},
"CreateStacks": {
},
"DynamoTiltSeriesAlignment": {
},
"DynamoCleanStacks": {
},
"BatchRunTomo": {
"skip_steps": [4],
"ending_step": 6
},
"StopPipeline": {
},As the result of this segment TomoBEAR will create a folder structure with sub-folders for the individual steps, corresponding to the JSON file blocks. You can monitor the progress of the execution in shell and by inspecting the contents of the folders.
Note
Upon success of an operation the fileSUCCESSis written inside of each folder. If you want to re-run a particular step, you can terminate the process, change parameters, remove theSUCCESSfile (or the entire sub-folder) and restart the process.
Since the input data already consists of the assembled tilt stacks, we did not need to setup:
- sorting individual raw frames/movies into corresponding tilt series, which can be performed by
"SortFiles": {}module inTomoBEAR; - beam-induced motion correction, which can be performed by
"Motioncor2": {}module inTomoBEAR.
The key functionality at this step is performed by TomoBEAR modules performing gold fiducials picking and tracking for subsequent tilt series alignment (TSA):
-
"DynamoTiltSeriesAlignment": {}module wraps the fiducials picking and (initial) tracking routines from the Dynamo package (a recommended Dynamo TSA tutorial); -
"DynamoCleanStacks": {}module excludes tilt stack projections containing low number of tracked gold beads; -
"BatchRunTomo": {}module creates IMOD project and imports previously obtained Dynamo-tracked fiducials into that project.
After IMOD project has been created and TomoBEAR stopped its automatic execution, you may proceed with inspecting the fiducials model in the folder, corresponding to the step "BatchRunTomo": {}, which you should be able to find in your processing folder (located in output/ sub-folder of the path, set up earlier in the parameter "processing_path").
For that, move to the corresponding folder:
cd /path/to/your/processing/folder/output/5_BatchRunTomo_1and open all tomograms in etomo in order to inspect and, if needed, refine initial fiducials tracking:
etomo tomogram_*/*.edfFurther you should perform etomo fiducials refinement procedure, which is briefly outlined below and described in details in the section VII. Alignment of serial tilts in the original etomo tutorial.
When etomo starts, choose the Fine Alignment step (left column in etomo GUI) and then click on Edit/View fiducial model button to start 3dmod with the right options to be able to refine the gold beads. If you do not see the green circles - please go to the IMOD command window, Edit -> Object-> Type-> activate Scattered radio-button and increase the size of the circle (e.g. to 5).
Note
It is also quite helpful to use Bead Helper plugin, showing tracks of fiducials throughout all the tilts.
Before you start to refine, just press the arrow up button in the top left corner of the window with the view port in order to zoom in. To refine the gold beads click on Go to next big residual in the window with the stacked buttons from top to bottom and the view in the view port window should change immediately to the location of a gold bead with a big residual.
Now see if you can center the marker better on the gold bead with the right mouse button. It is important that you don't put it on the peak of the red arrow but center it on the gold bead. When you are finished with this gold bead just press again on the Go to next big residual button. After you are finished with re-centering the marker on the gold beads you need to press the Save and run tiltalign button.
After you finished the inspection of all the alignments, you can start TomoBEAR as previously in the MATLAB command window by
runTomoBear("local", "/path/to/empiar10064_config.json")TomoBEAR should now detect that it has stopped at the previous step StopPipeline and continue its automated execution up to the next StopPipeline section. The following excerpt from the configuration file describes the next TomoBEAR steps:
"BatchRunTomo": {
"starting_step": 8,
"ending_step": 8
},
"GCTFCtfphaseflipCTFCorrection": {
},
"BatchRunTomo": {
"starting_step": 10,
"ending_step": 13
},
"BinStacks": {
},
"Reconstruct": {
},
"StopPipeline": {
},This segment starts with another "BatchRunTomo": {} block, producing aligned tilt stack according to the fiducial model, refined on the previous step.
Further, the module "GCTFCtfphaseflipCTFCorrection": {} provides functionality for estimation of the global per-tilt defocus value (by estimating Contrast Transfer Function model) using GCTF/CTFFIND4. You can inspect the quality of the estimated CTF model fit by going into the folder 8_GCTFCtfphaseflipCTFCorrection_1 and typing imod tomogram_xxx/slices/*.ctf and making sure that the Thon rings match the estimation. If not - play with the parameters of the GCTFCtfphaseflipCTFCorrection module.
One more "BatchRunTomo": {} section performs subsequent CTF-correction by phase flipping using Ctfphaseflip from IMOD, based on defocus value estimated in the previous section and listed in the corresponding *.defocus files.
Then binned aligned CTF-corrected stacks are produced by "BinStacks": {} and tomographic reconstructions are generated by "Reconstruct": {}. Both steps will be performed for the data at the all binning (down-sampling) levels listed in the "general": {} section (e.g. "binnings": [2,4,8]).
Proceeding with the execution (with runTomoBEAR(...) command as before) for the following group of steps will perform one of the particles localization methods - template matching (TM).
"DynamoImportTomograms": {
},
"EMDTemplateGeneration": {
"template_emd_number": "3420",
"flip_handedness": true
},
"DynamoTemplateMatching": {
"sampling": 15,
"size_of_chunk": [464, 464, 100]
},
"TemplateMatchingPostProcessing": {
"cc_std": 2.5
},
"StopPipeline": {
},The additional section "DynamoImportTomograms": {} produces files needed to be able to import all the generated tomograms in Dynamo Catalogue tool.
First, the corresponding 80S ribosome average map is fetched from EMDB and re-scaled to the proper voxel size by "EMDTemplateGeneration": {}.
Then the "DynamoTemplateMatching": {} section performs template matching procedure, which produces cross-correlation (CC) volumes as the results, which can be inspected (located at X_DynamoTemplateMatching_1/tomogram_xxx/tomogram_xxx.TM/cc.mrc). The selection of the binning level of the tomogram to be used can be controlled by the parameter "template_matching_binning" in the "general": {} section.
Note
At a high binning level (e.g. 8 or 16) using the whole volume as a single chunk is much more optimal than doing several chunks, so it is important to set the corresponding parameter"size_of_chank"to the size of the binned tomogram used for template matching.
Finally, in the section "TemplateMatchingPostProcessing": {} the highest cross-correlation peaks, over 2.5 standard deviations (as controlled by parameter "cc_std") above the mean value in the CC volumes, are selected, resulting in the set of coordinates of the particles (subvolumes) to be extracted. 3D particles being extracted and their corresponding coordinates and orientations are stored in the particles_table folder as a "*.tbl" file in the Dynamo table format.
In the section below you will find subtomogram classification projects that should produce you a initial subtomogram average map.
The first one is the multi-reference alignment project with a so-called true particle class and noise trap classes to first classify out false-positive particles produced by template matching, this happens at the binning which was used for template matching. In the end of the segment you should have a reasonable set of particles in the best class.
"DynamoAlignmentProject": {
"iterations": 3,
"classes": 4,
"use_noise_classes": true,
"use_symmetrie": false
},
"StopPipeline": {
},After subtomogram classification project is done, you should have a reasonable set of particles in the best class which you should select manually. To select the best class you need to go into the last DynamoAlignmentProject folder before the last produced StopPipeline folder, and then go to alignment_project_1_bin_y/mraProject_bin_y/results/iteQQQQ/averages (where iteQQQQ corresponds to the pre-last iteration folder) and type imod average_ref_CCC_ite_QQQQ.em to open produced average for each class CCC to identify the best class to use further (one should look like some structure, other ones would contain only noise).
Variables binning y, pre-last iteration number QQQQ and class numbers CCC can depend on parameters used in DynamoAlignmentProject. Foe example, this can be the folder 23_DynamoAlignmentProject_1/alignment_project_1_bin_4/mraProject_bin_4/results/ite0012/averages where you may find files average_ref_001_ite_0012.em, average_ref_002_ite_0012.em, and average_ref_003_ite_0012.em corresponding to the produced averages for 3 classes, from which you should choose the best one.
Once you have selected the best class, insert corresponding class number in the list [] as a value of the parameter "selected_classes" to the following section to be executed by TomoBEAR:
"DynamoAlignmentProject": {
"iterations": 3,
"classes": 3,
"use_noise_classes": true,
"use_symmetrie": false,
"selected_classes": [1],
"binning": 4
},
"DynamoAlignmentProject": {
"classes": 1,
"iterations": 1,
"use_noise_classes": false,
"swap_particles": false,
"use_symmetrie": false,
"selected_classes": [1],
"binning": 4,
"threshold":0.8
},
"StopPipeline": {
},The second section above is called single reference project, which will split the particles of the previously selected best class into two equally sized classes (called even/odd halves) with subsequent alignment of the particles in those halves to produce corresponding averages. This division will be needed further when unbinned data will be produced to be able to calculate the resolution of the resulting averaged map using Fourier Shell Correlation (FSC) curve.
After the first single reference project introduced above you will need to process tomograms by similar projects but at lower binnings in order to reduce the voxel size up to unbinned data to get the information corresponding to the highest possible resolution to be achieved using the current data set. At this point automated workflow is finished as the user needs to play with the masks, particle sets, etc.
You may want to try to use the following example of the end section of JSON file in order to have experience of processing tomograms at lower binnings to produce unbinned data to finally be able to calculate resolution of your ribosome electron-density map as a result of the first experience with TomoBEAR!
"DynamoAlignmentProject": {
"classes": 1,
"iterations": 1,
"use_noise_classes": false,
"swap_particles": false,
"use_symmetrie": false,
"selected_classes": [1,2],
"binning": 2,
"threshold":0.9
},
"BinStacks":{
"binnings": [1],
"use_ctf_corrected_aligned_stack": false,
"use_aligned_stack": true,
"run_ctf_phaseflip": true
},
"Reconstruct": {
"reconstruct": "unbinned"
},
"DynamoAlignmentProject": {
"classes": 1,
"iterations": 1,
"use_noise_classes": false,
"swap_particles": false,
"use_symmetrie": false,
"selected_classes": [1,2],
"binning": 1,
"threshold":1
}Consider, that after performing first single reference project you need to select both halves from the previous step by setting "selected_classes": [1,2] (in order to keep all particles) while producing one class by setting "classes": 1 at the subsequent steps of binning reduction using "DynamoAlignmentProject": {} module.
If you get out of memory error while running some of "DynamoAlignmentProject": {} at lower binnings (especially the last one), you may put additional parameter "dt_crop_in_memory": 0 to the corresponding "DynamoAlignmentProject": {} sections in order to prevent keeping the whole tomogram in memory for processing. For example, in this tutorial size of the one of unbinned tomograms is ~72Gb, while for binning 2 it is near 9Gb.
Finally, to estimate resolution of produced by TomoBEAR results, you need to use the following Dynamo command in MATLAB:
fsc = dfsc(path_to_half1, path_to_half2, 'apix', 2.62, 'mask', path_to_mask, 'show', 'on')
where path_to_half1 and path_to_half2 are paths to the prelast iteration results of the last DynamoAlignmentProject folder, which in this tutorial are located in 29_DynamoAlignmentProject_1/alignment_project_1_bin_1/mraProject_bin_1_eo/results/ite0006/averages, where you may find files average_ref_001_ite_0006.em and average_ref_002_ite_0006.em corresponding to the averages made from halves of the resulting particles set.
You also need to use a mask to filter averages for FSC calculation, and the accuracy of the used mask have impact on the resolution estimation. Appropriate mask to use for the initial resolution estimation you may find in the last DynamoAlignmentProject folder in a file called mask.em (in this tutorial path_to_mask is 29_DynamoAlignmentProject_1/mask.em).
After that you should get a similar FSC curve to the following one:
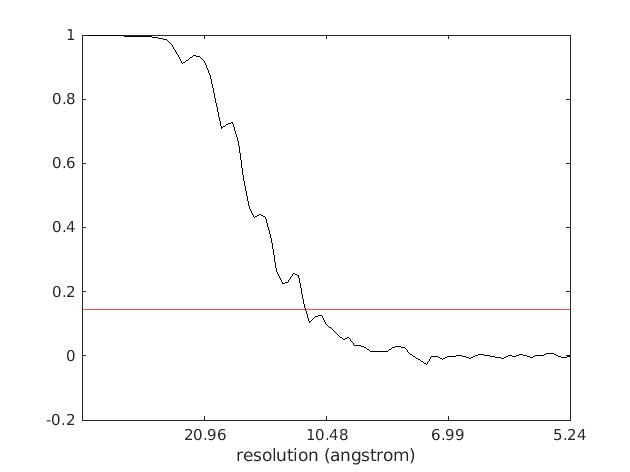
where in red we added a so-called "gold-standard" criterion of FSC = 0.143 to estimate the final map resolution, which in our case for the final set of ~4k ribosome particles reached 11.3Å.
Here the Ribosome data set-based tutorial is finished. We thank you for trying out TomoBEAR and hope you have enjoyed it!
This tutorial is devoted to the cryo-ET dataset containing in situ ryanodine receptor type 1 (RyR1) in SR vesicles, publicly available as EMPIAR-10452. This data set contains fiducials and consists of motion-corrected tilt stacks in ST (essentially MRC) file format.
In the TomoBEAR source code folder you will find a subfolder tutorials/empiar10452_ryr1 containing the following files:
-
empiar10452_download.sh- bash script to download raw data and gain files; -
empiar10452_config.json- configuration file to process this dataset with TomoBEAR.
As the second data set to showcase the capabilities of TomoBEAR we have chosen the plasma-FIB-milled data set EMPIAR-11306.This data set is fiducial-less and contains raw dose-fractionated movies in the Electron-Event Representation (EER) format.
In our case we performed EER movies integration using MotionCor2 utilities, than we used IMOD-based patch-tracking in order to align this fiducial-less data set with the following CTF-estimation and correction and IMOD-based reconstruction. Finally, we used Dynamo-based template matching to pick particles.
Note on StA part Further StA processing happened outside of the TomoBEAR pipeline because of the tools used and a lot of manual intervention needed to accurately process this data set at the StA processing stage, for details see our preprint: Balyschew N, Yushkevich A, Mikirtumov V, Sanchez RM, Sprink T, Kudryashev M. Streamlined Structure Determination by Cryo-Electron Tomography and Subtomogram Averaging using TomoBEAR. [Preprint] 2023. bioRxiv doi: 10.1101/2023.01.10.523437
In the TomoBEAR source code folder you will find a subfolder tutorials/empiar11306_ribo_pfib containing the following files:
-
empiar11306_download.sh- bash script to download raw data and gain files; -
empiar11306_imod_config.json- configuration file to process this dataset with TomoBEAR using IMOD patch-tracking for fiducial-less tilt-series alignment; -
empiar11306_aretomo_config.json- an alternative configuration file to process this dataset with TomoBEAR using AreTomo for fiducial-less tilt-series alignment; -
empiar11306_isonet_model_config.json- an additional group of configuration sections to perform missing wedge reconstruction and denoising (including pre-processing, training and prediction) using IsoNet; -
empiar11306_TM_steps_config.json- an additional group of configuration sections to perform template matching on this dataset.
This file describes the processing pipeline which should be setup by TomoBEAR to process the suggested for this exercise data set.
Using the outlined approach we were able to achieve 6.2Å in resolution with ~18.3k final particles. The resolution from the original publication by authors of this data set reached 4.9Å. We suggest you to try to process this challenging data set, starting from the example configuration file given below. We find it as a useful exercise for both novice and advanced TomoBEAR users to improve the suggested workflow template to achieve better resolution.