How to create new processing profiles
The GMS has entities called "processing-profiles" which specify the parameters required for running an analysis. For example, the number of threads for alignments, mismatch penalties, base quality cutoffs etc. Processing profiles help to maintain reproducible analysis results. This tutorial runs through the steps needed to create a new processing-profile by modifying existing processing-profiles. It is divided into multiple sections depending on the model-type (i.e. different pipelines).
- How to modify a Reference Alignment processing-profile
- How to modify a Somatic Variation processing-profile
- How to modify an RNA-seq processing-profile
##Reference-Alignment ###How to create a reference-alignment processing profile with modified read aligner parameters The reference-alignment model is typically used to align reads of a sample to a reference genome and call germ line mutations, calculate coverage reports, etc. In this example lets try to create a modified processing-profile for a reference alignment model.
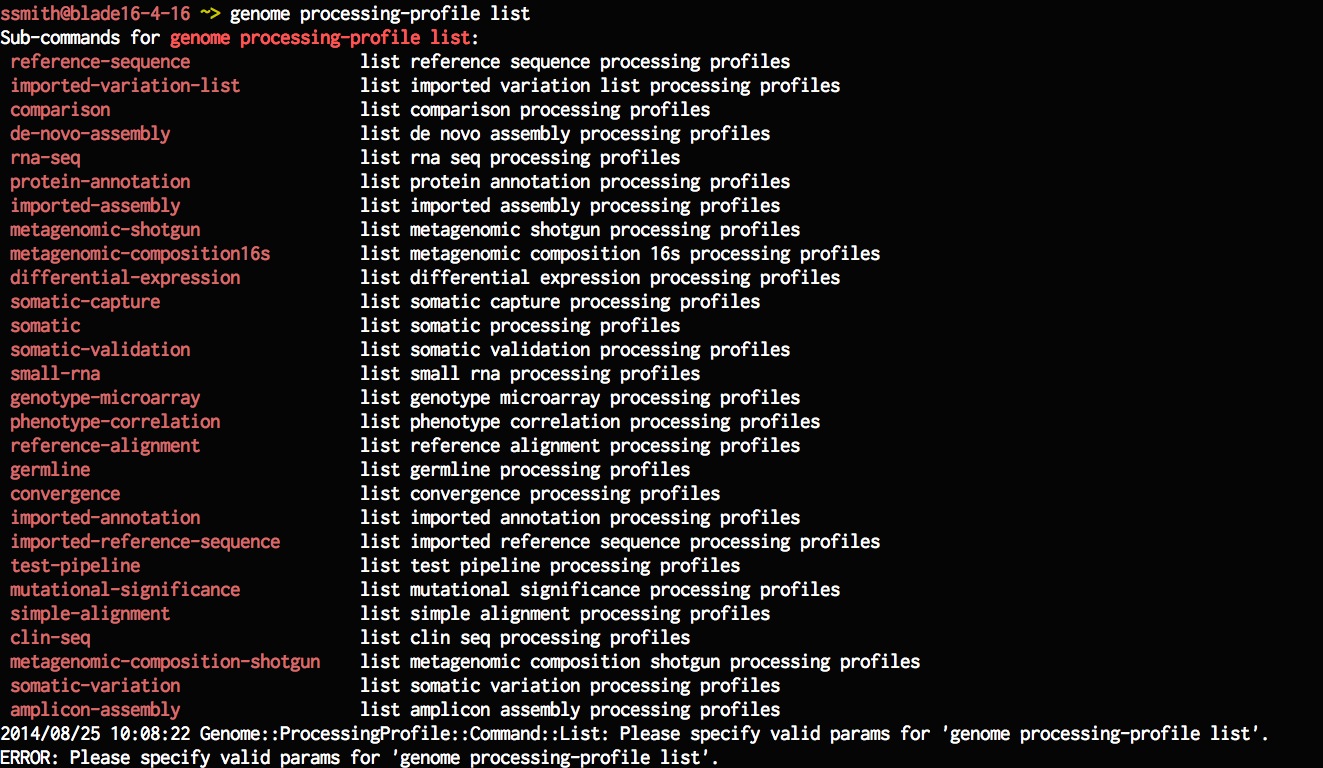
First list all the processing-profiles for ref-align models.
genome processing-profile list
genome processing-profile list reference-alignment
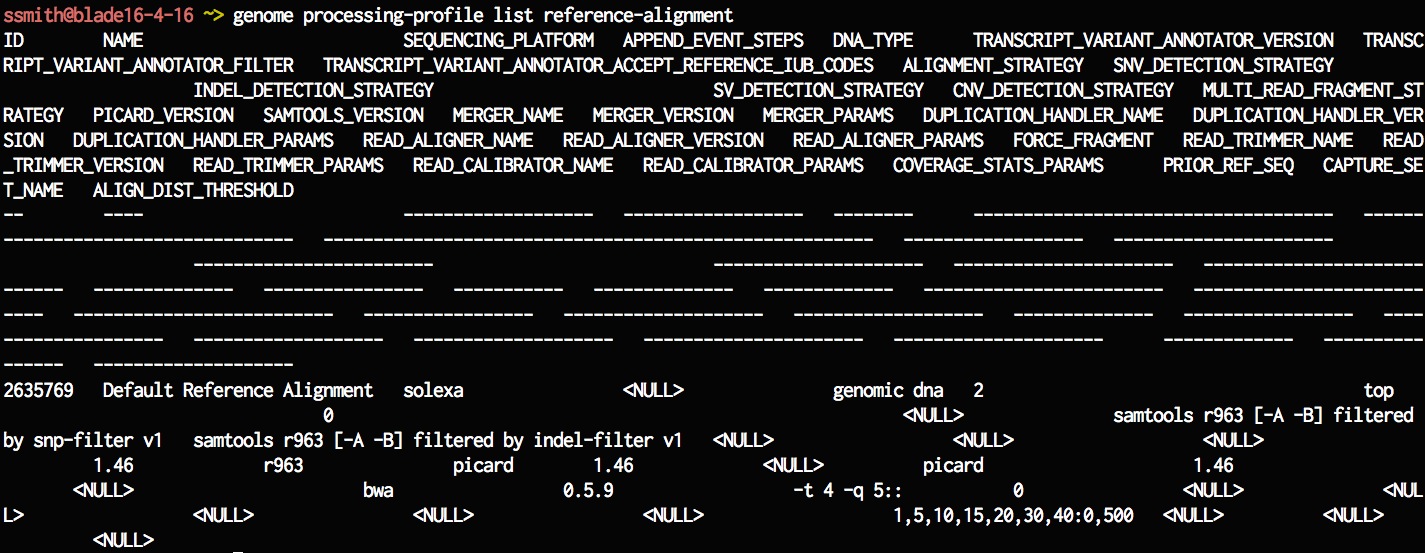
Lets try and look at the details of one of the ref-align processing-profiles. You can see the different parameter values using this command.
genome processing-profile describe 2635769
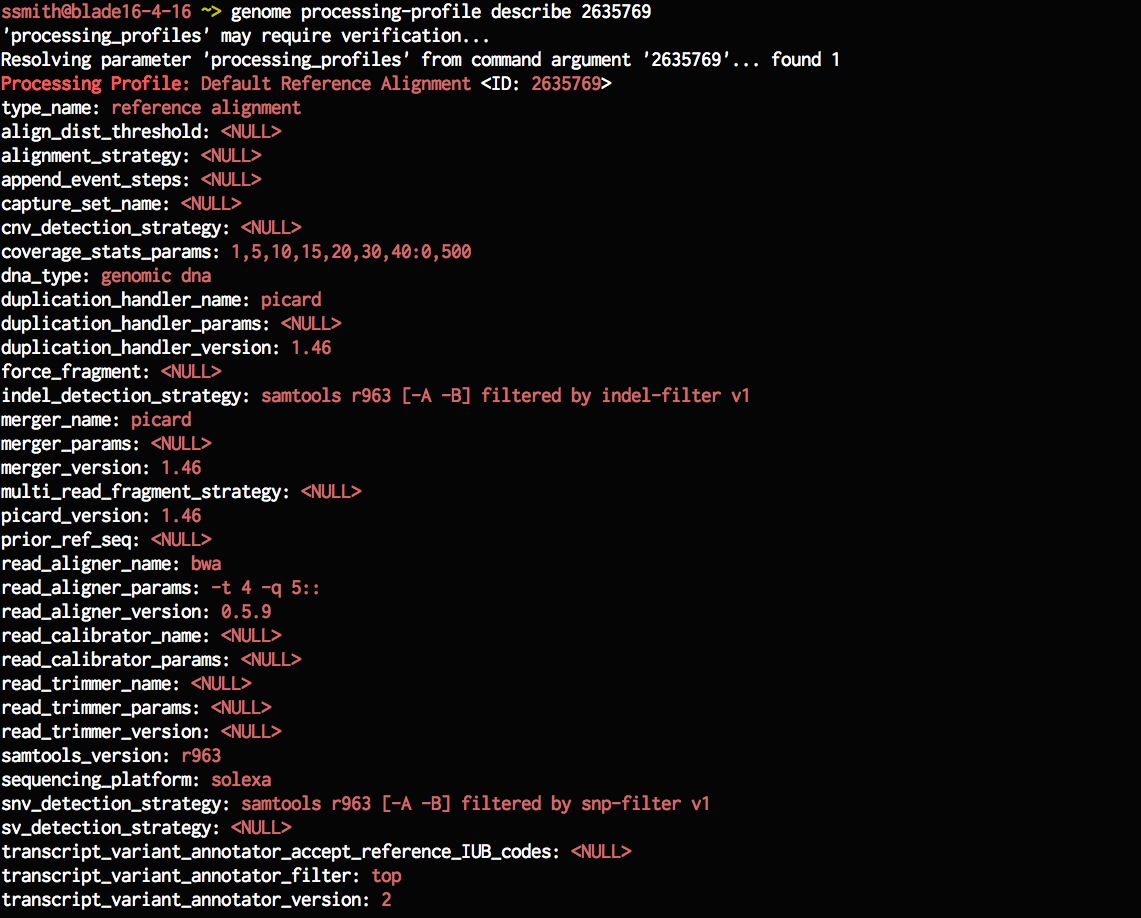
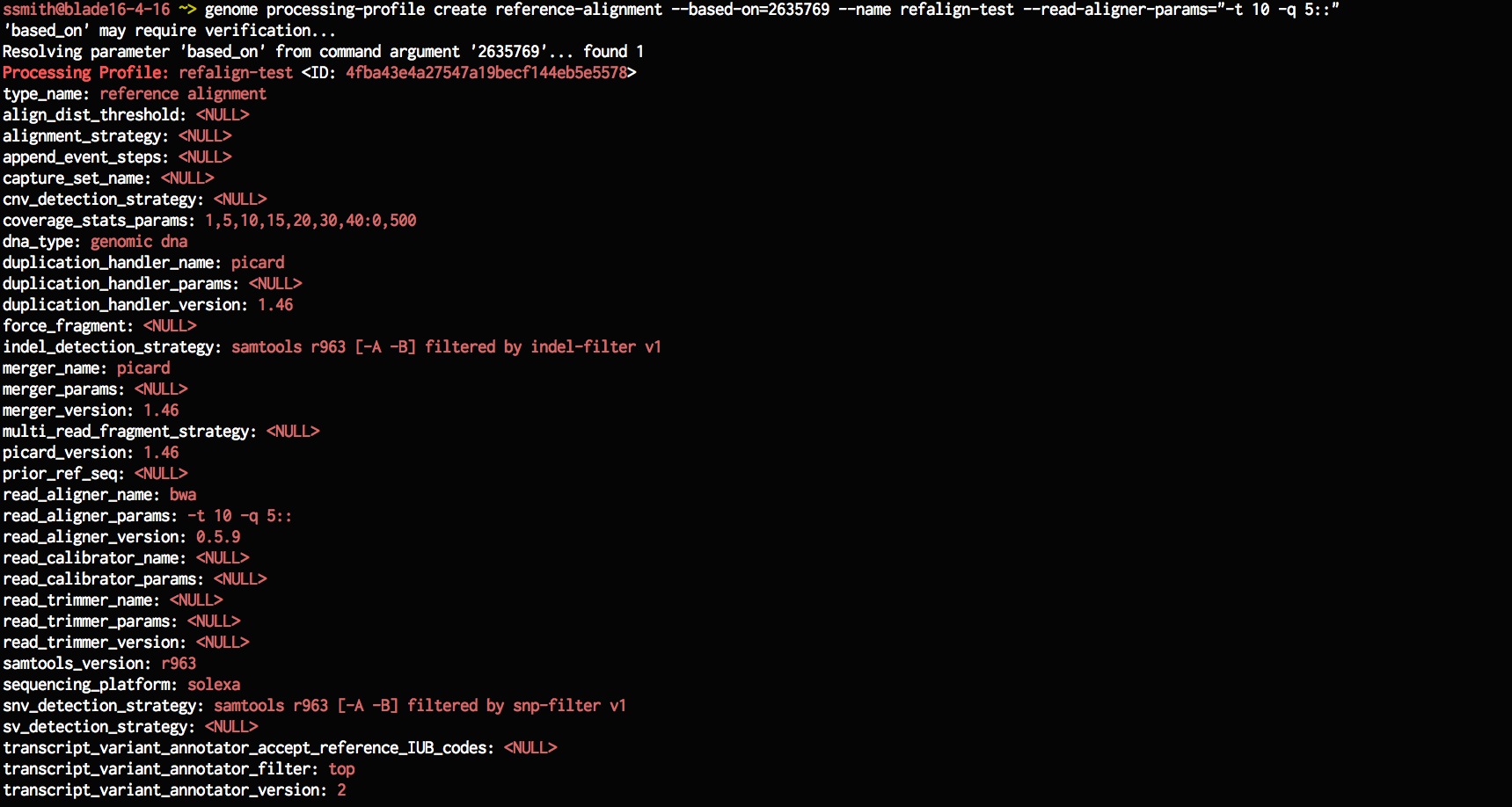
Lets now create a new processing-profile that is based on an existing processing-profile but has different parameters. Here, I've changed the read-aligner-params to use 10 threads instead of 4 threads from before. Once we've created the processing profile, list the available processing profile to check if the newly added one is in the list.
genome processing-profile create reference-alignment --based-on=2635769 --name refalign-test --read-aligner-params="-t 10 -q 5::"
genome processing-profile list reference-alignment

As a sanity check lets describe the new processing profile and see if it shows the desired changes.
genome processing-profile describe refalign-test
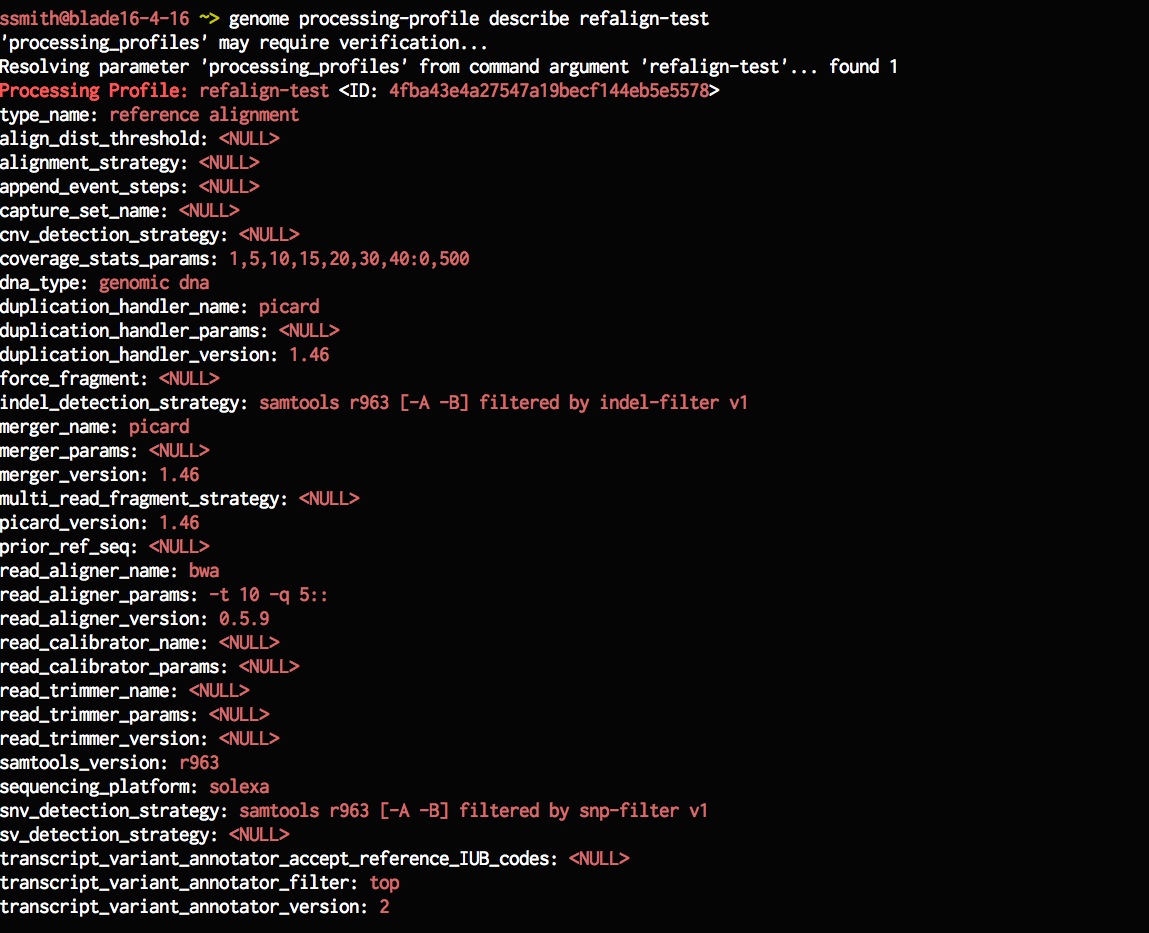
Now that it looks like we have a new processing profile, lets create a new model that uses it. Instead of creating a model from scratch lets try and create one from an existing model and just change the processing profile. This will copy the rest of the properties from the existing model, such as instrument-data, subject info, etc. and just update the processing-profile. Genome analysis doesn't always have to be hard does it?
Lets first list all the available reference-alignment models,
genome model reference-alignment list

Now lets copy an existing model to use the new processing-profile. Once we are done, lets list the models again to see the new model in the list.
genome model copy 2891377997 name=refalign-hcc1395-faster processing_profile=name=refalign-test
genome model reference-alignment list


Now its time to see that model in action and start aligning some reads. To do this we need to launch a build of this model like this,
genome model build start refalign-hcc1395-faster

Once the build is complete you can check the results in the data-directory of the build !
Similar steps are listed below with screenshots for updating the processing profiles of Somatic Variation models and RNA-seq models. As always if you have any questions, shoot us an email or even better, open a new issue on the GMS GitHub page!
##Somatic Variation ###How to create a WGS somatic-variation processing profile that drops the SV detection step Somatic Variation models are usually used to compare normal and tumor samples and call variants specific to the tumor. This is one of the standard workhorse pipelines of the cancer analysis groups at the Genome Institute. In this example lets look at how we can create a modified processing profile for a somatic variation model based on an existing processing profile. Scroll down when ready !
Lets first list all the available somatic-variation processing-profiles
genome processing-profile list somatic-variation
Lets have a look at the WGS somatic-variation processing profile in detail.
genome processing-profile describe 2762562

Lets create a new somatic-variation processing profile based on the existing one, lets remove the SV detection step from this one.
genome processing-profile create somatic-variation --based-on='2762562' --name 'wgs_somatic_variation_no_sv' --sv-detection-strategy ''

Confirm that the new processing-profile was created by listing it.
genome processing-profile list somatic-variation
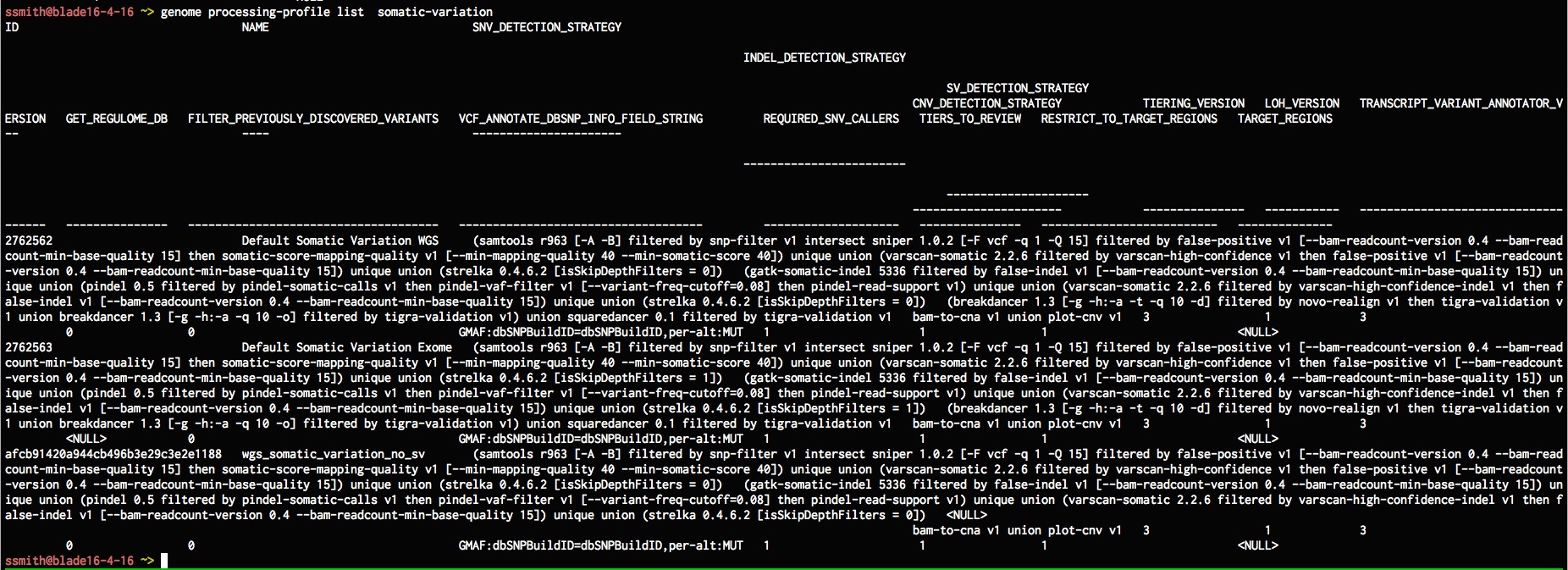
Now lets set up a somatic-variation model to use the new processing profile, lets list all the existing somatic-variation models.
genome model somatic-variation list
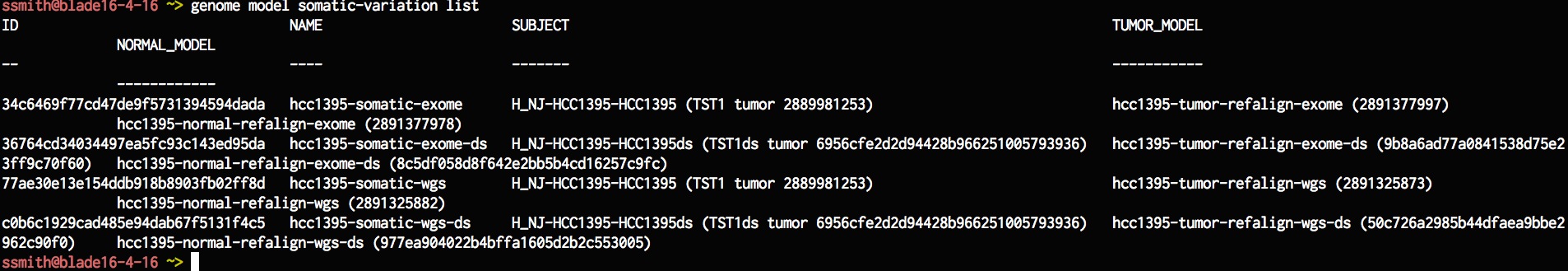
Lets create a new somatic variation model based on an existing model, everything is the same as the existing model except for the processing profile. For the processing profile lets specify the new one that we just created. Once we are done lets list the model that we just created.
genome model copy c0b6c1929cad485e94dab67f5131f4c5 name=hcc1395-somatic-wgs-no-sv processing_profile=name=wgs_somatic_variation_no_sv
genome model somatic-variation list


To actually see the model in action run a build of the newly created model. Once the build is done, the results will be in the data-directory of the build.
genome model build start hcc1395-somatic-wgs-no-sv

##RNA-Seq ###How to create an rna-seq processing profile that uses additional CPUs for alignment and expression estimation RNA-seq models are used to analyze RNA samples. These models calculate an estimate of transcript expression, find splice junctions, detect gene fusions amidst other analysis steps. In this example lets look at how to create an modified RNA-seq processing-profile.
Lets first list all existing processing-profiles.
genome processing-profile list rna-seq

Lets describe one of the processing-profiles in detail.
genome processing-profile describe 2762841
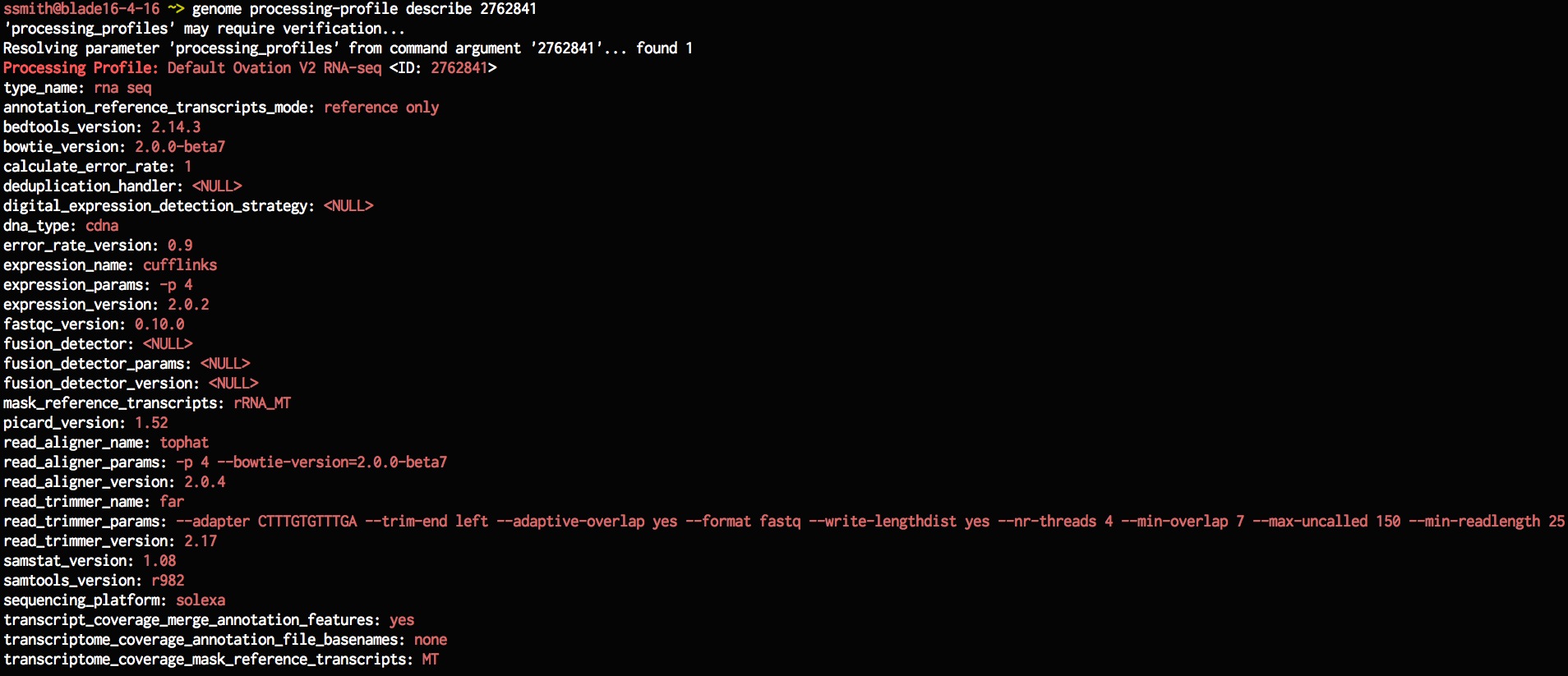
Lets create a new processing profile based on the existing one, lets change the aligner params to use 8 CPUs and lets also increase the number of CPUs used for estimating expression.
genome processing-profile create rna-seq --based-on='2762841' --name 'rnaseq_faster' --read-aligner-params '-p 8 --bowtie-version=2.0.0-beta7' --expression-params '-p 8'

Now we need to create a model to use this new processing-profile. Lets first list all existing RNA-seq models,
genome model rna-seq list
Lets create a new RNA-seq model based on the existing one but using the new processing profile. List the models again to verify that the new model was added.
genome model copy hcc1395-normal-rnaseq-ds name=hcc1395-normal-rnaseq-ds-faster processing_profile=name=rnaseq_faster
genome model rna-seq list


Now lets start a build of the model. When the build finishes running the results will be in the data-directory of the build !
genome model build start hcc1395-normal-rnaseq-ds-faster

To check the status of a build use
genome model build view $build_id
where $build_id is the ID of the build. For more useful gms commands refer to this page. If you've setup the GMS correctly you will receive an email when your build succeeds with the necessary logs(the email address to receive the reports is usually specified with $GENOME_USER_EMAIL.)
Good luck with your analysis !