Summary of results from the Clinseq pipeline
#Results from Clinseq This page is a summary of the results produced by the Clinseq pipeline of the Genome Modeling System.
In the examples below the base directory assumed is,
$clinseq_build_data_directory/$sample_identifier/
The $sample_identifier can be a individual_common_name like "BRC38", "TST1" etc or a sample_name like "H_KA007" or any other valid identifier depending on the samples that serve as input to the Clinseq model.
-
QC reports
-
Single Nucleotide Variants(SNV)
-
Indels
-
Copy Number Variation(CNV)
-
Loss of heterozygosity(LOH)
-
Structural Variants(SV)
-
Clonality
-
IGV sessions
-
Circos plots
One of the first things to look at when a clin-seq build is run for the first time is the metrics from the sequencing experiment. Things such as the amount of coverage observed in the various samples, the inferred insert size, concordance based on different metrics(e.g SNP concordance with dbSNP) etc. can provide valuable clues as to what to expect from the analysis results.
The QC reports are present in the input folder. The file QC_Report.tsv has a majority of the metrics that we typically used. This file lists the instrument-data, processing-profiles, input-builds, somatic-variation statistics, library level information such as insert-sizes and a bunch of other miscellaneous QC metrics. If you need them further metrics are available within the various subfolders under input. For example the wgs, exome and rnaseq folders contain per-lane and summary BAM-QC results for the different datasets. This can be useful to identify lane-level artefacts and capture efficiency for example. A lot of these metrics are produced by running the BAM files through Picard. Flowcell level information is also available under the APIPE_reports folder for different samples. This is produced by querying the APIPE databases with the sample name. We used to pull metrics from the LIMS tables as well but that process has hence been discontinued. A lot of the commonly used metrics are aggregated in the summary/Stats.tsv This file along with QC_Report.tsv typically contains most of the information that one needs to understand the quality of the sample/sequencing experiment.
The Single Nucleotide Variant(SNV) results are a major portion of the Clinseq build's output and there are several different directories containing these results currently. This can be quite confusing at first but once you identify your use-case you should be able to figure out where to look pretty quickly.
A couple of rough guidelines that might be helpful in this regard,
- If you are looking for read-support for all somatic variants in the sample look at SNV-indel-report This recently added tool should satisfy most of your needs with respect to SNVs and INDELs.
- If you are looking read-support information for previously known canonical mutations in cancer look at SNV-indel-report
- If you are looking for general patterns of somatic mutation such as mutation-rates or frequency of different nucleotide changes(A->C, G->T etc.) look at Mutation spectrum
- If you are looking for visualizations of the mutation within the context of a gene(the classic 'lolliplot') look at Mutation diagrams/Lolliplots
The SNV-INDEL report is perhaps the most widely used result from the ClinSeq pipeline. These are somatic SNVs and INDELs called by different variant callers and stored in the snv_indel_report folder. Most of the files in the folder contain headers which are hopefully self-explanatory. We usually look at variants that are called by multiple variant-callers and then manually review those.
Some notes about the different files in the folder are listed below.
- The
*unfiltered*version of the files contains all the variants - The
*filtered*version of the files contains the variants filtered by various filters, these are described in the perl module here, at the time of writing the different filters applied to the somatic variants were- min 20x coverage in both the tumor and normal sample.
- min 2.5% tumor variant allele frequency.
- min 3 variant supporting reads in the tumor sample.
- max 10% variant allele frequency in the normal sample.
- max GMAF of 2.5 at this locus. The GMAF is usually obtained from dbSNP. Please refer to the Perl module for more detailed explanations.
- The
*coding*files in this folder contain SNVs and INDELs that are not of the followingtrv_types
-|3_prime_flanking_region|3_prime_untranslated_region|3_prime_untranslated_region|5_prime_flanking_region|5_prime_untranslated_region|intronic|silent|rna|splice_region
The trv_type is the variant-annotation from the GMS annotator. The GMS annotator is an in-house annotator which can be invoked with gmt annotate transcript-variants.
- The
*clean*files in this folder have the same variants as the 'not-clean' equivalent, they just don't have the following columns -qw (gene_name transcript_species transcript_source transcript_version transcript_status c_position ucsc_cons domain all_domains deletion_substructures transcript_error gene_name_source)The clean versions were created in the hope that these are easier to read with the most frequently used columns.
The Database of canonical mutations(DOCM) is a resource that is an effort from folks at MGI. The web-server is here. We are obviously interested in using this resource as well. The way we use DOCM is by looking at all the DOCM positions and counting the number of reads supporting the reference and variant alleles using bam-readcount. This offers us to be very sensitive and look for variants that might have very little read support. This tradeoff of specificity for sensitivity is affordable since we are looking at a fairly 'small' number of curated sites. Almost all of these sites will have zero reads supporting the alternate allele.
DOCM has a concept of 'primary' and 'permuted' sites. The primary sites are the canonical mutations that were curated. For example BRAFV600E. We are also interested in nucleotide changes that result in changes to the neighboring amino acids, so we basically permuted the +/- 5 bases of the canonical sites to get the permuted sites.
So if the original ‘primary’ site was chr1:10000, we look for all possible changes from chr1:9995-10005
The column primary is a 0/1 field that tells you if this was a primary site or a permuted site.
In our experience so far we usually don't see reads supporting the permuted variants(most sites have zero reads supporting the variant allele) but it’s good to make sure. The primary sites are definitely interesting for followup.
The DOCM results are in the docm_report/ folder.
##CNV
Whole genome data usually produces the 'cleanest' CNV calls due to the uniform coverage profiles across the genome.
The algorithm we use for calling somatic copy-number changes is CopyCat by Chris Miller et al. The WGS cnv results are in the folder cnv/wgs_cnv.
The read-depth and CNV information for 10kb windows across the genome are in the wgs_cnv/cnvs.hq file. The format of this file is chromosome position tumor_readdepth normal_readdepth copy_number_difference_tumor_normal The copy-number segments identified by CopyCat are in wgs_cnv/cnview/CNView_All/cnaseq.cnvhmm.tsv These segments are of the format chr start end num_probes num_probes tumor_copynumber tumor_copynumber normal_copynumber normal_copynumber NA Gain/Loss As you can see, some of these columns are repetitive and one of the columns is actually NA, this is the format that the visualization tool CnView expects(based on an earlier copy-number caller called cnvhmm), and we've modified the pipelines to produce files of similar format.

The visualizations of the copy number calls are in the folder CNView_* These visualizations were produced by an in-house script called CnView. The y axis is the absolute copy number difference between tumor and normal. The background data is obtained from the cnvs.hq file while the actual copy-number variable segments are plotted using the cnaseq.cnvhmm.tsv. The color coding for gains and losses is done using absolute cutoffs which are indicated in the legends. The different folders CNView_All/, CNView_AntineoplasticTargets_DrugBank/, CNView_CancerGeneCensusPlus_Sanger/ and CNView_Kinase_dGene/ differ in the genes that are annotated in the figures. For example if you would like figures which highlight just the cancer genes the plots in CNView_CancerGeneCensusPlus_Sanger/ would be the ones to look at. This is useful especially for large segments when the number of genes in the identified CNV segment is large.
The copy-number results from Exome sequencing if available are in the cnv/exome_cnv/ folder.
The cnmops.cnvs.txt file contains the copy number calls. The first three columns are the identical to the BED format ("chr", "start", "end"). The next two columns are the median and mean copy-number difference between the tumor and normal copy-number for each segment. This is the absolute copy-number difference and is NOT log-scaled. The last column is the copy-number of the tumor assuming that the normal is diploid. For example, CN3 means that the tumor has 3 copies of this segment. If you’d like to use a discrete copy-number output, the last column would work. The median of the segment would work if you want to be more precise(not rounded to the nearest integer).
A quick check to visualize the CNV segments are the cnmops.segplot_*pdf plots. There are chromosome level plots
as well as a whole-genome plot. The x axis is the chromosome co-ordinate and the y axis is log(tumor_copy_number/normal_copy_number). You can deduce that if the tumor and normal have identical inferred copy-numbers(CnMops does this using the read-depth information.) you get log(1) which is equal to zero. Thus this plot usually has the majority of the points clustered around zero with the CNV segments being outliers.

The figures in the CNView* folders provide alternate visualizations, however these plots were designed with Whole Genome data in mind and since exome data usually has a higher variance in terms of read-depth these visualizations are usually incredible noisy and not very useful. The cnmops.segplot_*pdf plots are first recommended for any inference.
An effort was made to extract posterior probabilities for the CNV calls, CnMops did not provide this output by default at the time of writing. These values are in the cnmops.cnvs.filtered.txt file. However the version of CnMops currently installed at MGI seems to have a bug which prints sometimes spits out incorrect chromosomes in the cnmops.cnvs.filtered.txt This has been fixed in the newer versions of CnMops but we haven't switched over to the newer versions yet. Hence this file is not recommended to be used.
The exome-cnv step is run only on the regions which were captured for exome sequencing. The region of interest file is obtained from the reference-alignment model. This tells us what were the regions that were captured for sequencing. If the tumor and normal samples were run on distinct capture reagents then an intersection of the capture regions is used for the analysis. Using incorrect regions can render the exome-CNV analysis useless as it becomes hard to obtain an accurate picture of baseline normal coverage(skewed by regions of no/very low coverage). The actual genomic regions used for the analysis are saved in the file exome_cnv/tumor.normal.ROI.intersect.bed.
If you would like to debug what went wrong with the exome-cnv step or are super-inclined to dive into the data, the R session is stored as an R object in the file exome_cnv/cnmops.RD.Robject. Open up an R session and use load() to load this file into memory and you have all the data structures used to produced this result.
The microarray CNV results are in the cnv/microarray_cnv/ folder. The microarray data is probe level intensity data. CopyNumber is inferred by comparing the probe intensities to a set of reference probes and then by clustering(segmenting) probes with similar intensities. We use the Circular Binary Segmentation(CBS) algorithm by Olshen and Venkatraman for this analysis. This algorithm is implemented in the R package DNACopy. The segments with mean intensity above or below 'normal' intensity(determined from the reference probes) are inferred to be copy-number variable segments. When both tumor and normal microarray data is available the difference in the log intensity between the tumor and normal is used, when only the tumor microarray data is used analysis can be performed using just the tumor intensity data.
The individual probe level intensity and copy-number values are in microarray_cnv/cnvs.hq. The number of probes in this file depends on the array used. We currently use arrays which have about 4.2 million probes on them(most of these are common SNPs/INDELs).
The identified CNV segments are in the microarray_cnv/cnvs.diff.cbs.cnvhmm file. These segments are of the format
chr start end num_probes num_probes tumor_copynumber tumor_copynumber normal_copynumber normal_copynumber NA Gain/Loss As you can see, some of these columns are repetitive, this is the format that the visualization tool CnView expects(based on an earlier copy-number caller called cnvhmm).

The visualizations are in the folder CNView_* These visualizations were produced by CnView with WGS data in mind. The y axis is the absolute copy number difference between tumor and normal whereas microarray data is usually plotted on a log scale, hence these plots look incredibly noisy. The individual tumor and normal array files are in tumor.copynumber.original and normal.copynumber.original. These are obtained from the genotype microarray builds. These files contain LogR and BAF values at each probe for each sample.
##LOH ClinSeq attempts to identify regions of somatic LOH in the tumor.
###Brief workflow
The IdentifyLoh.pm module fetches unfiltered Varscan snvs from the somatic-variation build, these are then split by chromosome and then passed to FilterFalsePositives.pm which splits the SNVs into germline, somatic or LOH and filters out false-positives. The genome/lib/perl/Genome/Model/Tools/Varscan/ProcessSomatic.pm module splits the variants into somatic, germline and loh variants, this status is assigned by Varscan during variant-calling. These variants are then filtered by genome/lib/perl/Genome/Model/Tools/Somatic/FilterFalsePositives.pm the default filtering parameters are used in this module, the minimum variant allele read count is 4, and minimum variant allele frequency is 0.05, there are also other filters based on mismatch base qualities, proximity to end of reads, minimum strandedness etc.
The LOH and germline variants are then combined and segmented using Genome::Model::Tools::Varscan::LohSegments to identify segments of LOH. These segments are then filtered on segment-mean and number of SNVs in the segment. LOH segments with a average tumor variant allele frequency > 0.95 || < 0.05 (these are presumably homozygous in the tumor) AND with at-least 10 SNPs in the segment are retained. These two parameters are specified by the minprobes and min_segmean parameters of the IdentifyLoh.pm module.
Structural variants identified from DNA sequencing data are in the sv/ folder. Currently the structural variants are identified from WGS data using BreakDancer and a couple of validation tools such as Tigra-SV. Breakdancer calls SVs based on abnormal insert sizes between read-pairs. Modern SV callers such as Lumpy integrate information from split-reads as well but integrating Lumpy into the pipelines has been on our to-do list for a while now. The identified structural variants are in the file sv/svs.hq.merge.annot.somatic These structural variants have read-support in the tumor sample and no read support in the normal and hence potentially somatic.
The different types of structural variants that BreakDancer identifies are
- CTX - Inter-chromosomal translocations
- ITX - Intra-chromosomal translocations
- DEL - Deletions
- INV - Inversions
- INS - Insertions
The number of calls for each SV type are summarized in the file
sv/Stats.tsv. This is based on the raw calls prior to any sort of review.
Most of the focus in ClinSeq so far has been on inter-chromosomal translocations. These are denoted by the string "CTX" in the sv/svs.hq.merge.annot.somatic file. These are pulled out and stored separately in a different file CandidateSvCodingFusions.tsv.
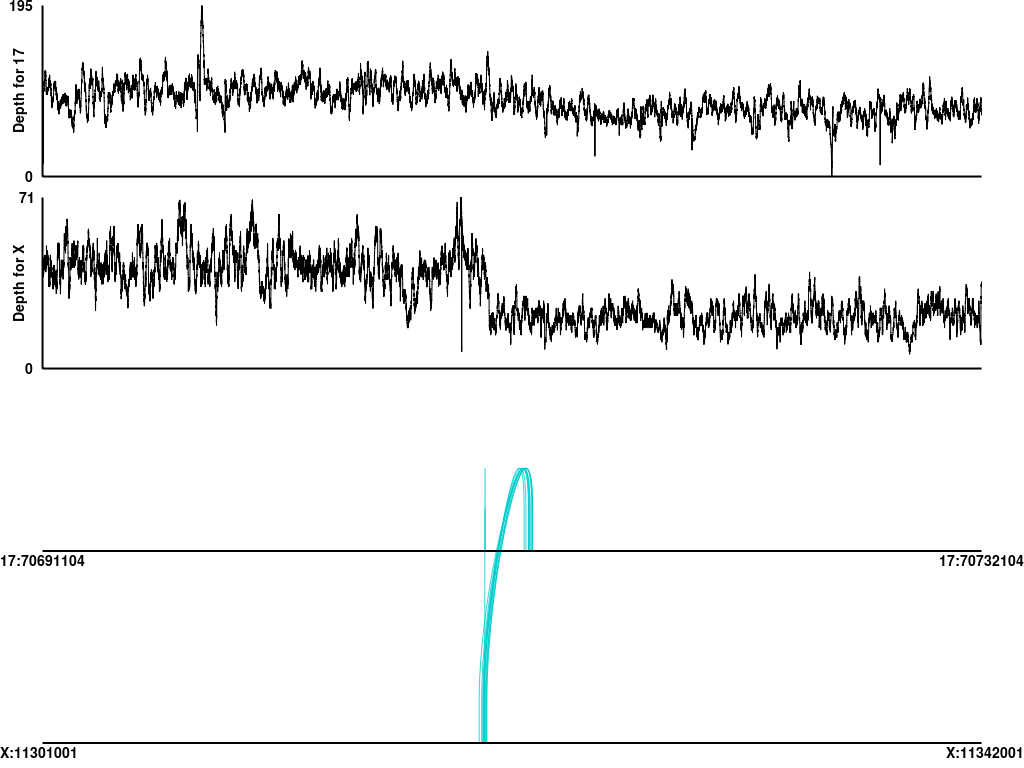
A basic visualization tool Pairoscope(http://pairoscope.sourceforge.net/) is run on the per-sample alignments to visualize the read-pairs which span the CTX junctions. These visualizations are stored in the sv/pairoscope folder. Separate visualizations are available for tumor and normal samples. The number of reads that Pairoscope finds is also stored in the CandidateSvCodingFusions.tsv file. SV's with Pairoscope read-support in both the tumor and normal are likely to be germline variants. These CTX's are also run through the DgiDB API to see if the SV spans any druggable targets. These results are in the sv/CandidateSvCodingFusions.tsv.dgidb/ folder.
Tumors we study are often heterogeneous, a mixture of cells with differing mutational histories. It is often important to understand the clonality of a tumor to guide treatment. Looking at the variant allele frequencies of somatic variants serves as a proxy for cell populations. While doing this analysis it is imperative to take into account the tumor purity, for e.g if there is not enough tumor cells in the sample that is sequenced even the variants in the dominant/initial clone appear at a lower variant allele frequency. It is also important to take the ploidy of the genomic region into account while looking at the variant allele frequency, we typically look at variants in the copy-neutral regions of the tumor as these are straightforward to analyze. Once we correct for purity and ploidy the variants can be clustered into different sub-clones. The computational method that we use for identifying the subclones is SciClone
The results from an earlier tool that we used for clonality are in the clonality folder. The SciClone results are in clonality/sciclone. The clonality pipeline is setup in such a way that the variant calling and copy-number results are fed into the clonality results. Also variants used in the clonality analysis are filtered using different minimum base and mapping quality cutoffs. The results using a base quality cutoff 20 and a mapping quality cutoff 30 can be found in the folder clonality/sciclone/b20_q30. Looking at the number of calls at different cutoffs gives us an idea about the quality of the data and the fidelity of the clonality analysis.
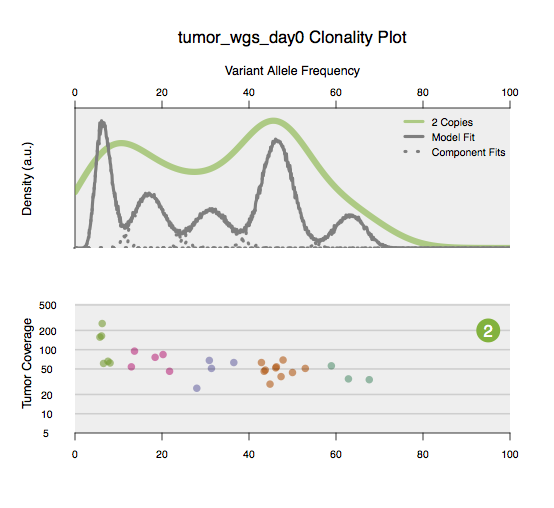
The variants used and the CNV results are also present in the same folder. The clonality analysis is run separately on WGS and exome data if both are available. The identified subclones are plotted in the file sciclone.tumor_exome_day0.clonality.pdf or sciclone.tumor_wgs_day0.clonality.pdf. The top panel shows the distribution of variant allele frequencies. The bottom panel shows the individual variants along with their coverage and variant allele frequencies. Separate panels are plotted for different ploidies. The ploidy is indicated by the number on the top right of the bottom panels, for example 2 indicates variants plotted in diploid regions.
The R script that replicates the SciClone analysis(sciclone.tumor_exome_day0.R) and the files containing the subclone information(sciclone.*.clusters.txt) are also present in the same folder. Whilst the automated analysis is a quick way to identify apparent clusters we typically use variants that we are really confident about and possibly manually reviewed to confirm the results. WGS data has a lot more events compared to exome data and we hence might be more powered to detect sub-clones in WGS.
It is always nice to pop open IGV and visualize/understand an event that algorithms have identified in the data. To make this process a little less painful session files are pre-generated and are present as XML files in the igv folder. All you have to do next is open IGV and click on Load session to load the session files.
The session files load the relevant alignments for the clinseq model, e.g WGS, exome and RNA seq alignments for the tumor and normal samples where available. You will see a bunch of session files numbered level1-level7, details about what variants/tracks are loaded in the different sessions can be found in the DumpIgvXml.log.txt file. The different BED files that are loaded are from the somatic-variation builds. Level1 auto-loads the minimum set of SNVs/INDELs.

Circos plots are useful to visualize most kinds of genomic variation in a single plot. Each source of variation can be plotted as a separate track/lines and based on the data available and events observed customized Circos plots can be created for any sample.
In Clinseq, an automated Circos plot is generated under the circos/ folder. The configuration file that was used to generate the Circos plot is named circos.conf.
A brief outline of the tracks plotted on a Circos plots is below, this is based on the figure above which is data generated from a breast cancer cell-line.
- The outermost track is an ideogram of all the chromosomes, genes with events in them(excluding amplifications/deletions, since these can be a lot of genes) are labelled on the ideogram.
- The next track contains the tier1 SNVs/INDELs plotted on a bar plot, each vertical line represents a SNV/INDEL. This track is plotted if WGS/Exome data is available.
- The next track shows the differential-expression status of genes. This is calculated as fold increase/decrease and plotted as a bar-plot. This track is only plotted when RNAseq data is available for both the tumor and normal sample. Genes with up-fold changes are in red/orange and the down-fold changes are in blue.
- The next track shows the copy-number deletions/amplifications. If WGS data is available, CNV calls from this data is used, else if exome data is available CNV calls from the exome data is plotted. The amplifications are in red/orange and the deletions are in blue.
- The innermost part of the Circos plot is used to depict translocations in the DNA and gene-fusions from RNAseq data. The lines connect different regions of the genome which are involved in the structural variants. The WGS structural variants are depicted by the black lines and the RNA seq events are depicted by orange/red lines.