- Introduction
- System Requirement and Dependency
- Installation Guide
- Typical Usages
- Example data in the package
- Tools in toolkit
- Results interpretation
- Contact Us
- Supplementary
Parallel-Meta Suite (PMS) is a visualized and interactive software package for microbiome analysis. It not only implements a comprehensive workflow that covers from sequence processing to chart plotting by state-of-art algorithms, but also provides an easy-to-use graphical interface for configuration and results interpretation, which is easy to startup for all-level users. The entire procedure of PMS has been optimized by parallel computing, enabling the rapid screening of thousands of samples. Both metagenomic shotgun sequences and 16S/18S/ITS rRNA amplicon sequences are accepted.
We strongly recommend that read this manually carefully before use Parallel-Meta Suite.
PMS only requires a standard computer with sufficient RAM to support the operations defined by a user. For typical users, this would be a computer with about 2 GB of RAM. For optimal performance, we recommend a computer with the following specs:
RAM: 8+ GB
CPU: 4+ cores, 3.3+ GHz/core
OpenMP library is the C/C++ parallel computing library. Most Linux releases have OpenMP already been installed in the system. In Mac OS X, to install the compiler that supports OpenMP, we recommend using the Homebrew package manager:
brew install gcc
For statistical analysis and pdf format output, PMS requires cran-R (http://cran.r-project.org/) 3.2 or higher for the execution of “.R” scripts. Then all packages could be automatically installed and updated by the PMS installer.
Vsearch has been integrated in the package. If you want to install/update manually, please download from https://sourceforge.net/projects/vsearch/ and put the “vsearch” to $ParallelMETA/bin/.
HMMER3 has been integrated in the package. If you want to install/update manually, please download from http://www.hmmer.org/download.html and put the “hmmsearch” to $ParallelMETA/bin/.
The latest released package:
For Linux/Win10 WSL, the source code package is here.
For MAC, the source code package is here.
PMS provides a fully automatic installer for easy installation.
a. Extract the package:
tar -xzvf parallel-meta-suite.tar.gz
b. Install
cd parallel-meta-suite
source install.sh
- Please “cd parallel-meta-suite” before run the automatic installer.
- The automatic installer only configures the environment variables to the default configuration files of “~/.bashrc” or “~/.bash_profile”. If you want to configure the environment variables to other configuration file please use the manual installation.
- If the automatic installer failed, PMS can still be installed manually by the following steps.
If the automatic installer failed, PMS can still be installed manually.
a. Extract the package:
tar –xzvf parallel-meta-suite.tar.gz
b. Configure the environment variables (default environment variable configuration file is located at “~/.bashrc” or “~/.bash_profile”)
export ParallelMETA=Path to Parallel-Meta
export PATH="$PATH:$ParallelMETA/bin"
export PATH="$PATH:$ParallelMETA/Rscript"
c. Then, active the environment variables
source ~/.bashrc
d. Install R packages
Rscript $ParallelMETA/Rscript/config.R
e. Compile the source code:
cd parallel-meta-suite
make
PMS provides a pipeline to automatically process the whole workflow.
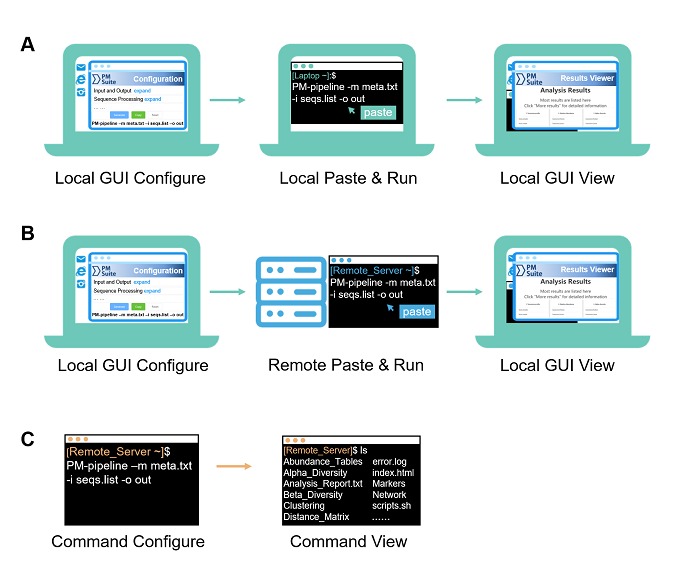
Fig. 1. Three typical usages of PMS for different scenarios and platforms. (A) Local GUI configure and run. (B) Local GUI configure and remote run. (C) Command configure and run (for both locally and remotely).
Local GUI-based usage is applicable with operating systems of Linux (GUI desktop installed), MAC OS, or Windows 10 (Subsystem for Linux (WSL) installed)
a. Open the "index.html" page in the GUI configuration folder ($ParallelMETA/PMS-Config/index.html) by a web browser.
Tip: You can also copy this folder to any other directory, e.g.
cp -rf $ParallelMETA/PMS-Config ./
b. Adjust parameters or keep the default options (according to actual requirements)
c. Click the “Generate” button at the bottom of the page to get a command
d. Click the “Copy” button to copy it into clipboard
a. Paste this single-line command in the local terminal, and it will perfectly run Parallel-Meta Suite pipeline without other operations.
a. Enter output directory
b. Open the visualized result viewer named as "index.html"
Tips:
- All original results (e.g. relative abundance table, distance matrix, etc.) are kept for further in-depth data mining or meta-analysis.
- In addition, the analysis summary, work log and detailed step-by-step workflow script are also provided in the result folder.
Usually, servers need remote login (e.g. via SSH) and only provide command-based terminal. in the local computer to generate the command.
a. Download GUI configuration guide folder ($ParallelMETA/PMS-Config/) from the remote server, and open "index.html" in the folder by a web browser
b. Generate the command in the local computer as well as local GUI Configuration
a. Paste and run the command in the terminal of remote server.
The results can also be transfer to the local computer for browsing as well as Local GUI View.
PMS also support command-based operations for non-GUI conditions, or experienced users. It is available via terminal either locally or remotely. Please run
PM-pipeline -h
to check the parameters of the automatic pipeline.
PMS package provides a demo dataset with 20 16S rRNA V4 amplicon-based microbiomes sequenced by Illumina platform. You can copy the dataset to the current path by
cp -rf $ParallelMETA/example ./
To run the demo, you can either
cd example
sh Readme
or use the GUI-based configuration
Then you can check the results by the visualized viewer index.html in the output folder by a webpage browser.
We also provide a demo output for this example dataset. Click here to download and check for details.
Tools can be directly used as Linux command line with parameters. To see all available parameters, please run the command with parameter ‘-h’, e.g.
PM-pipeline –h
| Command name | Purpose |
|---|---|
| PM-pipeline | Automatic analysis workflow. This tool integrates all other tools in this Table. |
| PM-format-seq | Sequence format check and reformation for PMS |
| PM-extract-rna | Extract amplicon fragments from shotgun sequences |
| PM-parallel-meta | Microbiome sample profiling |
| PM-select-taxa | Relative abundance table on selected taxonomy levels |
| PM-rand-rare | Samples rarefaction |
| PM-predict-func | Functional prediction |
| PM-predict-func-nsti | Compute the NSTI for predicted functions |
| PM-predict-func-contribute | Compute the contribution of taxon for predicted functions |
| PM-select-func | Relative abundance table on selected pathway levels |
| PM-plot-taxa | Taxonomy profile visualization by Krona |
| PM-rare-curv | Make the rarefaction curve |
| PM-comp-taxa | Compute the Meta-Storms distance among samples using taxonomy profiles |
| PM-comp-func | Compute the Hierarchical Meta-Storms functional distance among samples using function profiles |
| PM-comp-corr | Compute the correlation among community members, and construct the co-occurrence network |
| PM-split-seq | Split sequences by sample from merged sequences (e.g. QIIME format) |
| PM-update-taxa | Update the taxonomy annotation to the latest version |
| R-script name | Purpose |
|---|---|
| PM_Distribution.R | Plot relative abundances into bar-chat |
| PM_Adiversity.R | Alpha diversity indexes calculation and plotting |
| PM_Bdiversity.R | Beta diversity PERMANOVA and ANOSIM tests and plotting using distance matrix |
| PM_Hcluster.R | Hierarchical clustering by distance matrix |
| PM_Heatmap.R | Heatmap of distance matrix |
| PM_rarefaction.R | Rarefaction curve plotting |
| PM_Pca.R | PCA and plotting |
| PM_Pcoa.R | PCoA and plotting |
| PM_Marker_Test.R | Candidate biomarker selection by rand-sum test (for discrete meta-data variables) |
| PM_Marker_Corr.R | Candidate biomarker selection by regression (for numberic meta-data variables) |
| PM_Marker_RFscore.R | Biomarker ranking by Random Forest importance |
| PM_Network.R | Co-occurence network plotting using correlation among community members |
After using PM-pipeline, you might get the following folders/files in the output directory. In each directory, files/tables/figures are named with prefix “taxa” are taxonomy results, as well as “func” are metabolic functional results. PMS also provides an index page("index.html" in output directory) for results browsing.
This is the index page to browse for results browsing. Users can open it by a webpage browser and view the detailed results by hyperlinks.
This directory contains the visualized sample view in interactive pie charts across multiple samples.
This directory contains the relative abundance tables, absolute sequence count tables and bar charts of multiple samples on different taxonomical and functional levels.
This directory contains the pair-wised distance matrix of all input samples and unsupervised clustering results based on OTUs and KO profiles of multiple samples. Distances are computed based on Metastorms algorithm [Su, et al., Bioinformatics, 2012] and Hierarchical Metastorms algorithm [Zhang, et al., Bioinformatics Advances, 2021].
This directory contains the supervised clustering results based on PCA and PCoA.
This directory contains the multivariate statistical analysis results and rarefaction curve of alpha diversity.
This directory contains the multivariate statistical analysis results of beta diversity.
This directory contains the biomarker organisms and their Random Forest scores among different groups.
This directory contains the microbial interaction network based on different taxonomical and functional levels.
In this directory, each sub folder is the detailed information of an individual sample named by the sample ID. In the sub directories there may be
a. classification.txt (plain-text file): The OTUs and taxonomy information of this sample.
b. taxonomy.html (HTML webpage): The visualized sample view in interactive pie chart of this sample.
c. Analysis_Report.txt (plain-text file): The analysis report including parameters configuration and analysis information statistics.
d. meta.rna (fasta sequences): The extracted 16S/18S rRNA fragment, if the input is metageomic shotgun sequences.
This directory contains the profiling results path list of all samples. Each list has 2 columns: the first column is the samples’ ID and the second column is the path of the profiling result.
a. Analysis_Report.txt: The analysis report includes parameters configuration and analysis information statistics.
b. scripts.sh: The detailed scripts of each analysis step.
c. error.log: The warning and error messages.
Any problem please contact Parallel-Meta Suite development team
Chen Yuzhu E-mail: [email protected]
Li Jian E-mail: [email protected]
Dataset 1 contains 894 microbiome samples from a hospital's indoor environment before and after opening. [1] (size ≈ 1GB)
Dataset 2 contains 2,556 microbiomes sampled from multiple environmental conditions and studies. [2-9] (size ≈ 5GB)
Dataset 3 contains 14,000 microbiome samples from American Gut Project. [10] (size ≈ 9GB)
- Lax, S., et al., Bacterial colonization and succession in a newly opened hospital. Sci Transl Med, 2017. 9(391).
- Wu, G.D., et al., Linking Long-Term Dietary Patterns with Gut Microbial Enterotypes. Science, 2011. 334(6052).
- A, D.L., et al., Host lifestyle affects human microbiota on daily timescales. Genome biology, 2014. 15(7).
- Koenig, J.E., et al., Succession of microbial consortia in the developing infant gut microbiome. Proceedings of the National Academy of Sciences of the United States of America, 2011. 108.
- Muegge, B.D., et al., Diet Drives Convergence in Gut Microbiome Functions Across Mammalian Phylogeny and Within Humans. Science, 2011. 332(6032).
- Peiffer, J.A., et al., Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proceedings of the National Academy of Sciences of the United States of America, 2013. 110(16).
- Bulgarelli, D., et al., Structure and Function of the Bacterial Root Microbiota in Wild and Domesticated Barley. Cell Host & Microbe, 2015. 17(3).
- K, G.J., et al., Human genetics shape the gut microbiome. Cell, 2014. 159(4).
- Iratxe, Z., et al., The Soil Microbiome Influences Grapevine-Associated Microbiota. mBio, 2015. 6(2).
- Daniel, M., et al., American Gut: an Open Platform for Citizen Science Microbiome Research. mSystems, 2018. 3(3).