LAI does not improve despite sequencing and assembly improvements #87
Comments
|
Hi Weihan, Thank you for your detailed descriptions. I open a new issue because this is a different but interesting topic. We demonstrated in the LAI paper, that genome size, TE content, contig N50, BUSCO, etc, are not significantly correlated with LAI. Of course back then we only had a limited number of 'good' genomes to test on, and their quality was not evenly distributed. I have noticed similar cases but those are sequenced by Nanopore. I thought it may have something to do with the sequencing technique, but from your case it may be more prevalent. I am still collecting similar cases because so far it is sporadic and thus I have limited power to detect the cause. By the time I got enough data, the raw LAI correction may need a re-calibration. You mentioned assembling the genome with >20kb subreads, what is the coverage? Can you post one of the LAI screen outputs for this genome? Thanks, |
|
Hi Shujun, Thank you for your patience and help. The final assembly result is pretty good (reasonable genome size, low number of contigs, high contig N50). The LAI is 16.38. Below is my LTR_retriever log, I use LTR_FINDER and ltrharvest to find LTR, all parameters are default as you set: I observed that the number of intact LTR is about 100-300 less than that of other samples. I didn't save the LTR_retriever log of other samples, so I only show the *.LAI files of other sample. Please let me know if you need other information or LTR_retriever running logs of other samples, I can rerun LTR_retriever at any time. In additionally, you mean the raw LAI correction may need a re-calibration when you got enough high-quality genomes? Like 2.8138 or the equation?Maybe I can help you to collect some high-quality reference genomes that currently have a relatively high LAI. Thanks, |
|
Hi Weihan, Thank you for the feedback. Please run LAI independently and capture the screen output, or check the directory and find the .iden (or .age, forget which one) information. I suspect if this genome has less LTR activity and the LAI was not corrected well. Another good piece of information is to plot a histogram of intact LTR age with a couple of your genomes (with bin width as 0.2 MYA). The special genome has less total LTR and also less intact LTR in the assembly, I saw similar cases in Solanaceae species. For the correction yes it's the 2.8138 factor currently but it may need more corrections if we want to approach that route. It's hard to be fair with all species so careful evaluations are required. I suscept the length of LTR is also involved, so if you get a chance, you may also check if your genomes have different LTR length. Best, |
|
Hi Shujun, Thanks again for your patient help. Below is my LAI screen output: Below is my THG.rename.fasta.out.q.LAI.LTR.ava.age file: Below is the histogram of intact LTR age. Here I also share the simple plot script, hoping to help someone in need. Best, |
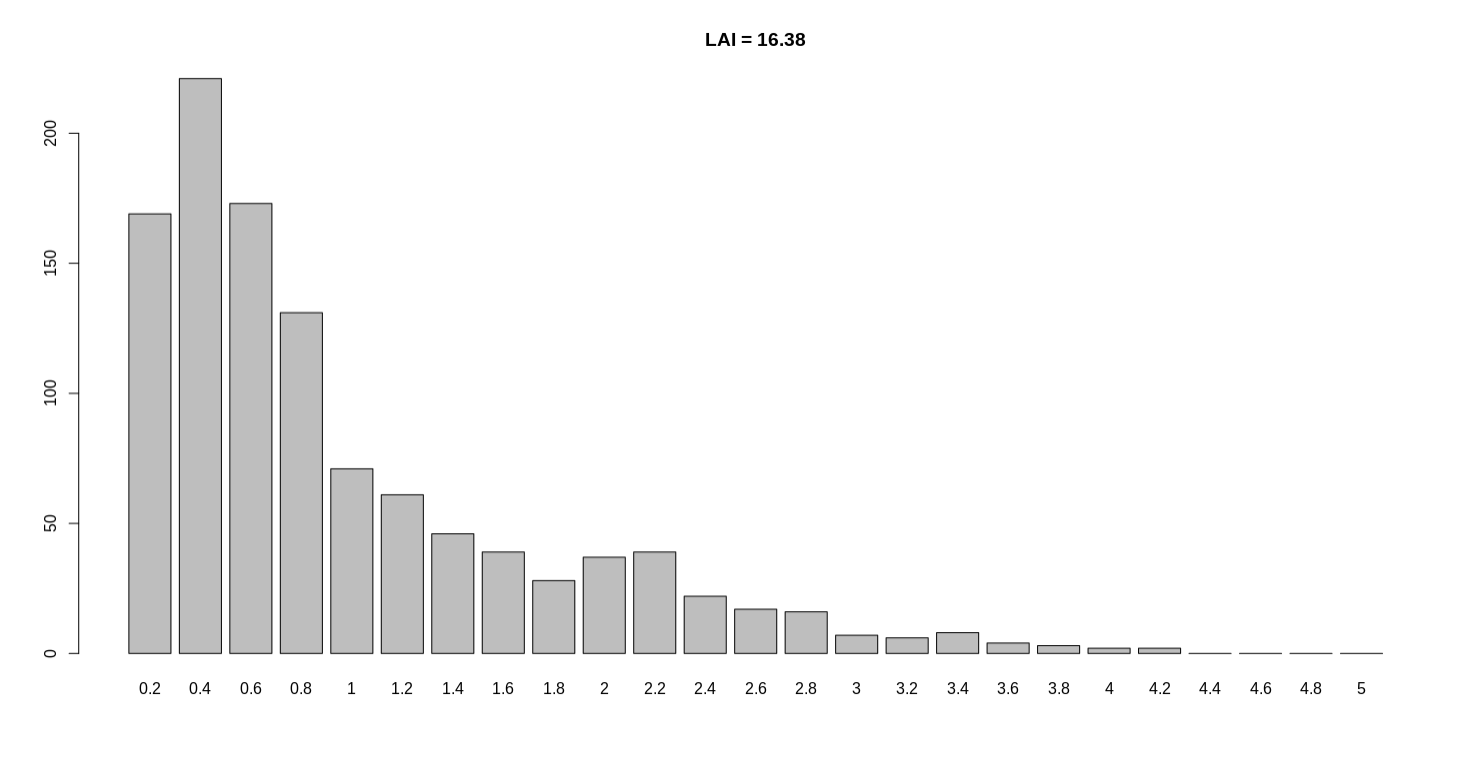
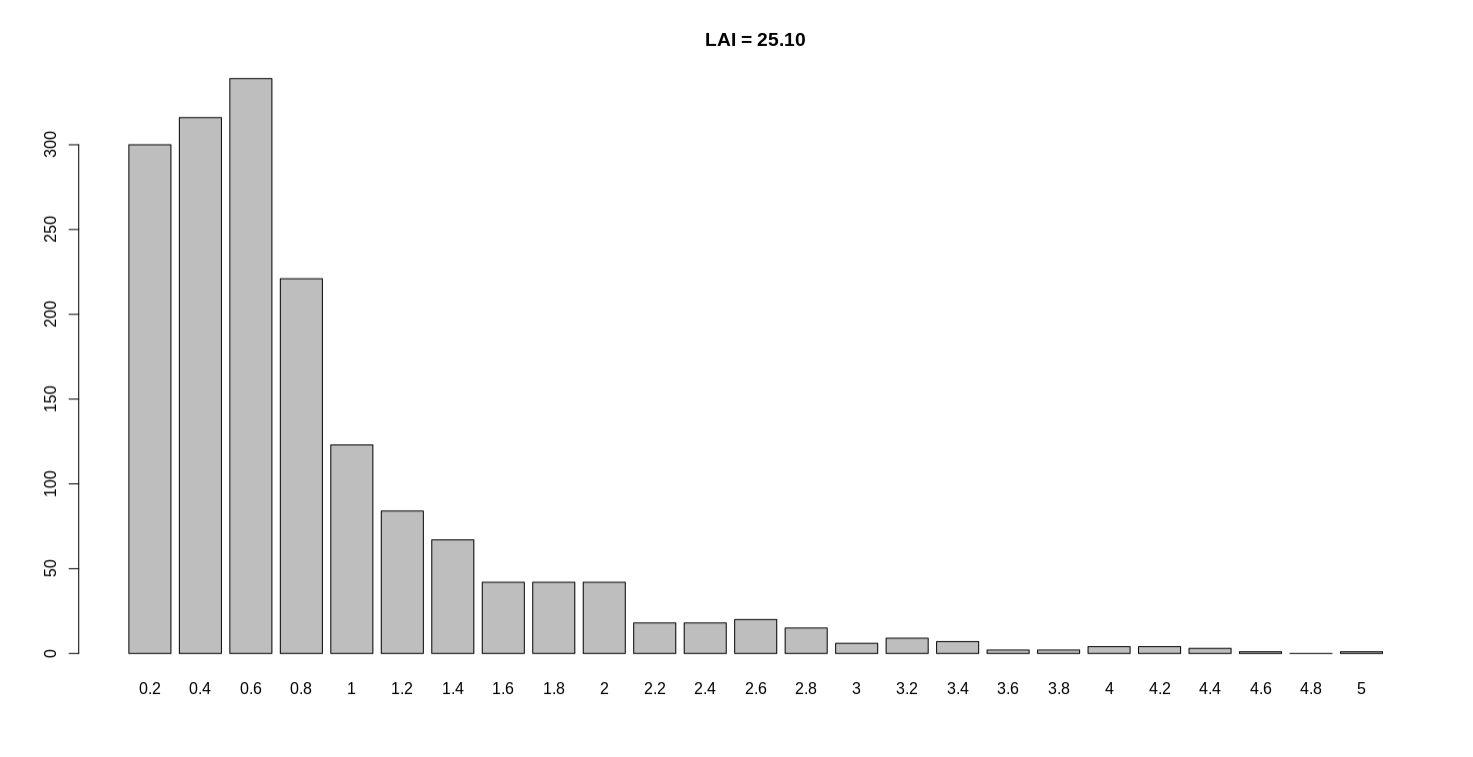
|
Hi Weihan, Thanks for sharing the data and scripts. It looks like your genomes have fewer young LTRs. Did you polish the genomes? Both Arrow and Pilon can fix sequencing errors and help to identify intact LTRs. Best, |
|
Hi Shujun, I tried to polish the genome these days by Arrow and Pilon. However, Pilon is slower and still running, so I used NextPolish instead. For only Arrow polish, LAI has slightly improved, from 16.38 to 16.40. For Arrow + NextPolish, LAI has been further improved from 16.40 to 16.48 The result did not look as ideal as expected. Do you have any other suggestions to improve LAI? Best regards, |
|
Hi Shujun, This is the LAI for Arrow (1 round) + Pilon (1 round) The 2nd round of Pilon is still running. It seems that polish can improve LAI. Do you have any suggestions for improving LAI? Best regards, |
|
Hi Weihan, Thanks for sharing the data. At this point, you have probably tried everything you can to improve the genome. It is likely that LAI can not evaluate this ancestral genome properly. You may state that in your manuscript. For research purposes, can you help to characterize the mean length of LTR regions from intact LTR-RTs in these genomes? In the pass.list file, you can find coordinates of these LTR regions. Thanks! Best, |
|
Hi Shujun, Thanks for your recent help. For the no polish genome LTR_retriever result (LAI = 16.38), the mean length of LTR regions is 6519.417. For the Arrow + Pilon polish (LAI = 17.61), the mean length of LTR regions is 6499.817. I calculated the length of LTR regions by the first column from pass.list file. E.g. For "Seq1:1526070..1531424", the length is 1531424-1526070. Best regards, |
|
Hi Weihan,
Thanks for getting these data quickly. LTR-RTs have the structure of
LTR-internal-LTR, So LTR region means either one of the LTR sequences.
There is a column IN:xxx-yyy, which is the coordinate of the internal
region, this will give you a way to get the coordinates of the left or
right LTR region. Can you also include a couple of the high LAI genomes for
this value? Thanks!
Best,
Shujun
…On Sat, Feb 6, 2021 at 4:46 PM Weihan ***@***.***> wrote:
Hi Shujun,
Thanks for your recent help.
For the no polish genome LTR_retriever result (LAI = 16.38), the mean
length of LTR regions is 6519.417.
For the Arrow + Pilon polish (LAI = 17.61), the mean length of LTR regions
is 6499.817.
I calculated the length of LTR regions by the first column from pass.list
file. E.g. For "Seq1:1526070..1531424", the length is 1531424-1526070.
Best regards,
Weihan
—
You are receiving this because you authored the thread.
Reply to this email directly, view it on GitHub
<#87 (comment)>,
or unsubscribe
<https://github.com/notifications/unsubscribe-auth/ABNX4NGF2ERGWYQ26ZEHPF3S5T6VBANCNFSM4WWLL2JA>
.
|
|
Hi Shujun, Thanks for your correction, the following is my recalculation results. For another genome LAI = 25.10, the mean length of LTR regions is 5400.613. (no polish) If you need other results, please feel free to contact me. Best regards, |
|
Hi Weihan, Thank you for the information. It seems to be a negative correlation between LAI and LTR region length, but it doesn't make sense on the equation. Supposely if the LTR is longer, the total intact LTR length is longer and raw LAI is higher. So I think length is not playing a big role here. I don't have other ideas at the moment. I will keep this issue open and if anybody has similar issues, they may report here. Thanks again for helping to diagnose the issue. Best, |
|
hi,Shujun Ou, The parameter of value 1 is set The parameter of value 2 is set Other parameters are the same as the official recommendation. But the difference between the LAI values obtained by the two methods is very large, so I want to know which result is the more reliable LAI value. From the distribution of LAI of each chromosome, I prefer to believe in low LAI. |
I saw this notice by just now when I had lunch, it looks very interesting... If you combine LTRharvest and LTR_FINDER, but just change LTRharvest Sorry I don't know if you understand what I mean. As I know, LTR_FINDER_parallele set the default Weihan |
|
Hi @chaimol, Weihan is correct, the total LTR content annotated via approach 1 is 39.75% and the second is 12.20%. While theoretically it's impossible to know the exact LTR content in your species, you may estimate it with annotations. Your two annotations are very different, thus making the LAI values very different. You need to make a decision on which LTR content estimation is more reliable. You may also need to combine LTRharvest and LTR_FINDER inputs to make a calculation more comparable with other genomes published with the LAI method. Best, |
Sorry, I did not clarify the specific parameter settings. In fact, value1 and value2 both use |
|
@chaimol It's your responsibility to determine which estimation of total LTR content is closer to the truth. If you think, for example, the total LTR content should be 50%, then you need to use Shujun |
|
Hi @oushujun , I'm following up on a topic I started in EDTA, but has to do with estimating LAI within species, sometimes between different assemblies of the same genotype: CLR vs HiFi, or different assemblers. Therefore, total assembly –and scaffolded– sizes can be quite different, which may or may not be relevant to my question. Below are the LAI values for the various assemblies of the same genotype, but notice that they are performed in scaffolded assemblies with different sizes: Following your advice, I added Here the new LAI values: Since a constant genome size parameter did not work, would it be valid or too unfair for the smaller assemblies to scale the Intact and Total percentages to the "real" genome size? In a way, I'm not too surprised that LAI values are not that different even between CLR and the best HiFi assembly. I have good evidence that the Megabases missing in the CLR assembly are mostly centromeres and rDNA clusters, and not that many contig breaks are due to TEs. This is why I have my doubts whether estimating LAI in the smaller assemblies with the parameters from the largest assembly. At the moment, I kind of favour the raw_LAI from the first list (without fixed Kindly, Fernando |
|
Hello Fernando, I have not pushed the As I stated in the LAI paper and also in the LAI output, raw LAI is suitable for within species comparisons while LAI works for both within and between species comparisons. However, raw LAI did not have genome size controlled. Having Judging on the results shared above, these assemblies are very close to each other in terms of TE assembly quality. You may want to look at other quality metrics such as N50, BUSCO, or assembly errors to further select your best assembly strategy/result. Best, |
|
Hi @oushujun, Thanks for pushing the Just for the purpose of completeness in this thread, I post here the LAI values of the same assemblies having controlled for In this case, it becomes apparent that the fixed genome size heavily penalises smaller assemblies. I'm not convinced these are the best parameter choices in this particular case, due to the fact that I know that what is missing in the smaller assemblies are mainly the core centromeres. One would have to make the assumption that these loci carry the same proportion of LTRs as the rest of the genome to make this extrapolation valid. As you've suggested, I'll complement these observations with other quality metrics. Thanks! |
|
Hi Fernando, Thank you for sharing your results. If you find the fixed genome size penalises smaller assemblies too much, it could be that the genome size is set too high. If the assemblies are smaller due to lacking some components of the genome, then the lower LAI value correctly reflects this. Centromeric regions usually harbor higher percentage of LTR sequences compared to whole-genome levels. Missing centromeres may contribute to lower assembly quality of LTR sequences genome-wide. If you want to compare the quality of the assembled part of the genome, you may want to extract the assembled part of all assembles based on synteny, then compute LAI on these sequences. Best, |
Dear shujun,
First of all apologize for my bad English. I have sequenced dozens species, these species belong to the same genus (eg. wild, cultivars, landrances), and I will construct a pan-genome. Sorry I am not an expert in sequencing experiments either. But I know they all use the same kit and extraction method to build the library.
Here I give you a detailed description of the only sample with LAI < 20. This sample named SampleA. At the beginning of my project, dozens of samples were all sequenced about 120 ~ 160X by Pacbio. The subreads length also well and subreads N50 are 10 ~ 20k. This looks really good so I assembled them by CANU and obtained dozens of genomes. The LAI value of all these genomes exceed 20 except SampleA is 16. And the contig N50 of SampleA also very unusual (just 200kb). So we contacted technical experts to perform another round sequencing. This time I got about 500X new Pacbio data of SampleA, also with normal subreads length and subreads N50. I assembled SampleA and run LTR_retriever, the contig N50 improved to ~5Mb while the LAI is still ~16. Surprised me and incomprehensible. Since I have enough data, I also tried only use length > 8kb, 10kb, even 20kb subreads to run different assembly software, all the LAI values is between 16 and 17, stable as Mount Tai. According to our common sense and your articles published on Nature communication, high-depth sequencing will improve assembly quality. The contig N50 is significantly improved (200kb to 5Mb), but the LAI value no improvement.
While writing here, I thought about it again. If it is a problem with library building, the contig N50 will not be improved. I used to think there was a problem with the DNA extraction process, human factors like some experimental operations. However, I got reasonable geome size, high contig N50 and high BUSCO (99%). So my guess last time about sequencing library construction may be unreasonable. The problem that still bother me is high-depth sequencing assembly get a long contig N50 but low LAI value. Different assembly methods and parameters have an effect on contig N50, but it doesn’t seem to have effect on LAI. Even I only use subreads length > 10kb to run assembly the LAI is still ~16. I have high-depth sequencing and long reads, the LAI hasn’t improved.
Thank you for taking the time to discuss so much with me.
Best regards,
Weihan
Originally posted by @Weihankk in #86 (comment)
The text was updated successfully, but these errors were encountered: