The next revolution in biology will be by computational biologists
In collaboration with Symbionts, Dept. of Biological Sciences, BITS Goa and SAiDL (Society for Artificial Intelligence and Deep Learning)
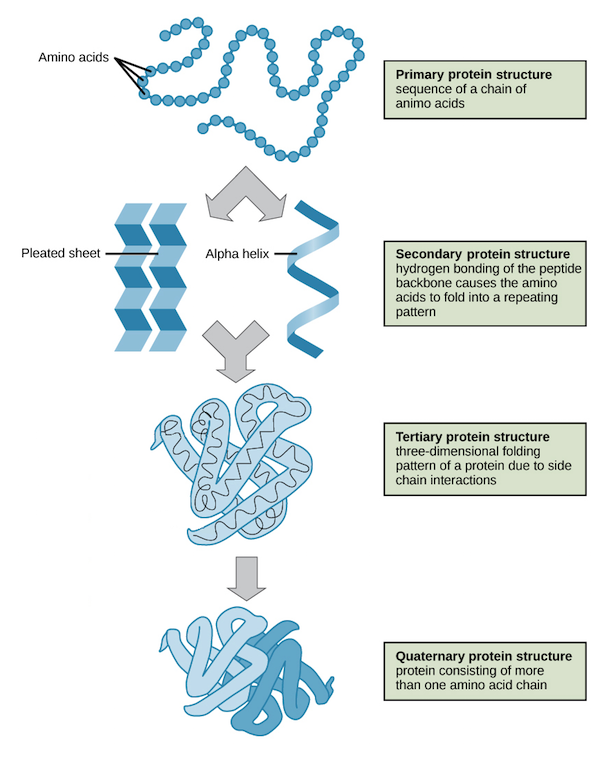
Protein sequencing is relatively much easier and cheaper to perform due to new technologies. But, with just a sequence we won't be able to determine the function of a protein. Protein structure determination is almost a necessary step in finding its function and even to engineer new proteins for varous applications. Several methods are currently used to determine the 3D structure of a protein, including X-ray crystallography, NMR spectroscopy, and Electron microscopy. They are extremely time consuming and expensive. Enter, Computational Biology and Machine learning. We need to build a Deep Learning model which takes a protein sequence (fasta format, see below) and gives a 3D structure (with coordinates of each amino acid in the protein, angles between the bonds, etc.
Amino acids are organic compounds that combine to form proteins. Amino acids and proteins are the building blocks of life. The 20 amino acids that are found within proteins convey a vast array of information. Each amino acid is represented as a single letter as shown below.

Example of an protein sequence with 330 amino acids (This is called the fasta format, in the realm of Computational Biology):
MKTAYIAKQRQISFVKSHFSRQLEERLGLIEVQAPILSRVGDGTQDNLSGAEKAVQVKVKALPDAQFEVVHSLAKWKRQTLGQHDFSAGEGLYTHMKALRPDEDRLSPLHSVYVDQWDWERVMGDGERQFSTLKSTVEAIWAGIKATEAAVSEEFGLAPFLPDQIHFVHSQELLSRYPDLDAKGRERAIAKDLGAVFLVGIGGKLSDGHRHDVRAPDYDDWSTPSELGHAGLNGDILVWNPVLEDAFELSSMGIRVDADTLKHQLALTGDEDRLELEWHQALLRGEMPQTIGGGIGQSRLTMLLLQLPHIGQVQAGVWPAAVRESVPSLL
{kind=link}
Several methods are currently used to determine the structure of a protein, including X-ray crystallography, NMR spectroscopy, and Electron microscopy. They are extremely time consuming and expensive. This is where Machine Learning and Deep Learning comes into the picture. Predicting 3D structure of protein from its amino acid sequence is one of the most important unsolved problems in biophysics and computational biology. Watch these interesting videos to gain more insight into the problem we are trying to solve: TedX: The protein folding problem, The protein folding revolution: Proteins and AI, BERTology: BERT meets biology and Khanacademy: Protein basics.
The NetSurfP-2.0 paper's model architecture is based on Bi-Directional LSTM. NetSurfP-2.0 implements the blossom matrix for feature extraction from the sequence of amino acids. But recent progress in the field of Natural Language Processing like Google's BERT has opened up better ways to extract features.

Task is to classify sections of a protein sequence into 3 secondary structures:
- H = 4-turn helix (α helix). Minimum length 4 residues.
- E = extended strand in parallel and/or anti-parallel β-sheet conformation. Min length 2 residues.
- C = coil (residues which are not in any of the above conformations).
Example (Corresponding result for the above sequence):
CCCCHHHHHHHHHHHHHHHHHHHHHHHCEEECCCCCEEECCCCCCCCCCCCCCCCEECCCCCCCCCEEECCCCCCHHHHHHHHCCCCCCCEEEEEEEEECCCCCCCCCCCCCEEEEEEEEEECCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCCCCCCEEEEHHHHHHHCCCCCHHHHHHHHHHHHCEEEEECCCCCCCCCCCCCCCCCCCECCCCECCCCCECCEEEEEEEECCCCEEEEEEEEEEECCHHHHHHHHHHHCCCCHHHCHHHHHHHCCCCCCEEEEEEEHHHHHHHHHCCCCHHHCCCCCCCHHHHHHCCCCC
BERTology Meets Biology, NetSurfP-2.0 and Evaluating Protein Transfer Learning with TAPE have implemented Transformers and Bi-Directional LSTMs for the classification task. For v0.1, we will be taking a lot of inspiration from these papers and come up with a combination of them. We will be taking the NetSurfP-2.0 as our baseline.
You have the choice to help improve our model and help fix issues. We would rather have you work on novel models (not limiting to NLP) and compete with our model! We will be updating pending work, issues on our model soon. Meanwhile, it is up to you to find more papers and models. Novel models are most welcome.
We are borrowing the dataset from DTU Bioinformatics Institute's NetSurfP-2.0. The training set is represented this way: Raw data is given in Numpy (Python) compressed files with an array of pdb/chain ids (pdbids) and a 3-dimensional array (of shape (10848, 1632, 68)) of input and output features. First dimension is protein samples, second dimension is sequence position and third dimension is input features. There are 10848 different protein sequences and largest sequence is 1632 amino acids long. And, each amino acid has the following data:
[0:20] Amino Acids (sparse encoding)
Unknown residues are stored as an all-zero vector
[20:50] hmm profile
[50] Seq mask (1 = seq, 0 = empty)
[51] Disordered mask (0 = disordered, 1 = ordered)
[52] Evaluation mask (For CB513 dataset, 1 = eval, 0 = ignore)
[53] ASA (isolated)
[54] ASA (complexed)
[55] RSA (isolated)
[56] RSA (complexed)
[57:65] Q8 GHIBESTC (Q8 -> Q3: HHHEECCC) ## We are replacing the different types of helices, Beta sheets from the OG dataset with just 3 classes - Helix, Beta sheet and Coil
[65:67] Phi+Psi
[67] ASA_max
We have prepared a starter kit on Colab to quickly get started with the dataset.