The human immunodeficiency virus (HIV) is a retrovirus that infects human cells, especially CD4+ T-cells. HIV infection is a dreadful disease that requires lifelong treatment. Although there have been developed several therapeutics that inhibit HIV replication, they are not curative. That's why dealing with this virus is an important task. HIV has several mechanisms for escaping human immune system: one of them is a high mutation rate. In this project we are to investigate the molecular evolution of HIV over time, changes in proteins of the HIV due to mutations and their relation to an escape from an immune system.
- Longitudinal HIV sequencing data was taken from https://hiv.biozentrum.unibas.ch/
- Representative proteomes were taken from https://www.uniprot.org/proteomes. For all taken proteomes UPIDs can be found in accession_id.txt.
High mutation rate of the HIV is aimed to accommodate virus to exact human immunity in order to escape it. HIV population typically mutates in the first few months after the infection. The mutations are thoroughly studied and documented, but little focus was placed on the protein level. It is known that these mutations are preferentially found in CTL epitopes. However, an interesting question is whether these mutations are directed toward self, i.e., if they are aimed at becoming more like a host proteome or not.
Main hypothesis of our project is that HIV mutates in direction toward its host to escape recognition. Although it's known that mutations of the HIV occur in order to escape immune answer but we assume that these mutations are also providing similarity to human proteins.
In order to validate the hypothesis we are going to measure the probability of HIV peptide sequences to be similar to a host (i.e. human). We derive the probability from the machine learning models trained to discriminate between human and pathogen peptide sequences.
First of all for each patient we've made evolution paths of the virus. For that reason single-patient phylogenetic trees for chosen region were created with pairwise alignments. Paths in those trees (from latest date to the reference) were taken as HIV evolution paths. Further analysis include translating DNA into peptide sequences and applying quantitative and qualitative methods to them.
In the quantitative analysis, frequencies of all possible amino acid k-mers (2-mers in our work) were calculated. To investigate the changes of similarity of HIV populations and self over time we trained a Random Forest model to discriminate between human and pathogen protein sequences. Then HIV evolution paths were used to validate our hypothesis.
In qualitative analysis translated sequences were used to calculate hydrophobicity metrics according to Kyle-Doolittle hydrophobicity score or Kidera factors. Obtained data was also combined with paths of the virus to test our hypothesis.
Pip and conda prerequisites can be found in pip_prerequisites.txt and conda_prerequisites.txt respectively.
It's recommended to use a computer with at least 2 GB RAM and 2.00 GHz CPU, especially while training machine learning models using our scripts.
Evolution trees: Phylogenetic trees for HIV haplotype were created using day-by-day structure - each level equals to one day of sequencing:

Trees tend to have one isolated path except for the last day of sequencing (or one day before last day). It can be interpreted as only one haplotype at each day was able to survive and become an ancestor for other haplotypes, however this interpretation may be incorrect. Further analysis should be carried out.
Quantitative analysis: In quantitative pipeline a classifier using 2-mers was trained on representative proteomes (see Data section). The accuracy of the classifier is about 84% in a split between human (class 1) and non-human proteins (class 0). Classification report:
| Class | Precision | Recall | f1-score |
|---|---|---|---|
| 0 | 0.86 | 0.84 | 0.85 |
| 1 | 0.83 | 0.85 | 0.84 |
Using quantitative pipeline on region V3 we have proved main hypothesis to be valid only for a part of the patients studied: patient p1, patient p4 (approximately), patient p11 (approximately). For other patients our model shows inconsistent results.
 For other regions results can be not so similar to V3 region, example for RRE region:
For other regions results can be not so similar to V3 region, example for RRE region:
 We need more data to extensively validate the hypothesis or to eventually reject it.
We need more data to extensively validate the hypothesis or to eventually reject it.
Qualitative analysis: For further analysis the longest path from the reference node in a phylogenetic tree was used (red edges). A tree for one region of a patient is shown below.
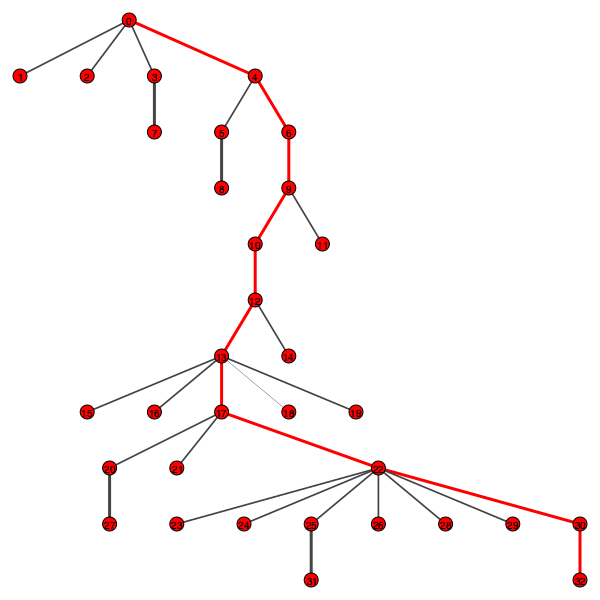

Qualitative analysis also can't provide clear answer on direction of HIV mutation.
Examples of how to use functions can be found in jupyter notebooks.
With our code you can make:
- Download data from this database
- Single-patient phylogenetic tree
- Train Random Forest on representative proteomes
- Use our explained pipelines
- Make a tool library
- Improve accuracy and consistency of our methods
- Include analysis for sequencing data from other papers about HIV
Pull requests are welcome. For major changes, please open an issue first to discuss what you would like to change.
Please make sure to supply issues and pull requests with tests.
We want to show our appreciation to:
- Fabio Zanini and Co. for making helpful interesting paper and giving free access to their database
- Bioinformatics Institute for additional help in creating our team
- Vasily Tsvetkov, Institute of Bioorganic Chemistry
- Alexandra Ovsyannikova, National Research Nuclear University MEPhI
- Andrey Kravets, Moscow Institute of Physics and Technology
- Lolita Alekseeva, Moscow State University
- Denis Khamitov, Moscow State University