Rootstrap
layout: userdoc title: "Rooting phylogenies" author: AUTHOR date: DATE docid: 8 icon: info-circle doctype: tutorial tags:
- tutorial description: Inferring rooted trees using non-reversible models. sections:
- name: Inferring unrooted tree with outgroup url: inferring-unrooted-tree-with-outgroup
- name: Inferring rooted trees without outgroup url: inferring-rooted-trees-without-outgroup
- name: Testing root positions url: testing-root-positions
Using time-reversible Markov models is a very common practice in phylogenetic analysis, because they provide high computational efficiency. However, these models infer unrooted trees hence lack the ability to infer the root placement of the estimated phylogeny. In order to compensate for the inability of these models to root the tree, many researchers use external information such as using outgroup taxa or additional assumptions such as molecular-clocks.
This guide provides the outgroup approach and another rooting approach using non-reversible models (Naser-Khdour et al., 2021), which will be useful when an outgroup is lacking. Please make sure that you use IQ-TREE version 2.1.3 or later for full features below and cite this manuscript:
S. Naser-Khdour, B.Q. Minh, R. Lanfear (2021) Assessing Confidence in Root Placement on Phylogenies: An Empirical Study Using Non-Reversible Models. https://doi.org/10.1101/2020.07.31.230144
We first demonstrate the outgroup approach to root the Bovidae family of five sampled species (Yak, Cow, Goat, Sheep and Tibetan antelope) using two outgroup species (Pig and Whale). Please download:
- An input DNA alignment file for these 7 species.
- An input partition file that defines 52 genes in this alignment. This is a subset of the mammal dataset (Wu et al., 2018).
Choosing a "good" outgroup is an entire topic on its own. In generally, the outgroup must contain taxa that do not belong to the ingroup but are evolutionarily close enough to the ingroup taxa.
To infer an unrooted tree, run:
iqtree2 -s bovidae_outgroup.phy -p bovidae.nex -B 1000 -T AUTO --prefix rev_dna_outg
that will invoke the ultrafast bootstrap with 1000 replicates (-B 1000), detect the
optimal number of threads (-T AUTO) and write all output files with the prefix rev_dna_outg.
The input alignment contains protein-coding genes. We can ask IQ-TREE to translate
the alignment into protein sequences using the standard genetic code (-st NT2AA) and perform
an amino-acid analysis on the translated alignment with:
iqtree2 -s bovidae_outgroup.phy -p bovidae.nex -B 1000 -T AUTO -st NT2AA --prefix rev_aa_outg
where setting the prefix to rev_aa_outg avoids file overwriting with the previous run.
The resulting tree may now look like (extracted from rev_aa_outg.iqtree):
NOTE: Tree is UNROOTED although outgroup taxon 'Yak' is drawn at root
Numbers in parentheses are ultrafast bootstrap support (%)
+--Yak
|
+--Cow
|
| +--Goat
| +--| (100)
| | +--Sheep
| +---| (100)
| | +---Tibetan_antelope
+---| (100)
| +-------------------------------Wild_pig
+----------------------| (100)
+-------------------Minke_whale
You can open rev_aa_outg.treefile in a tree viewer software (e.g. FigTree) and re-root
the tree on the branch separating the outgroup (Wild_pig and Minke_whale)
from the remaining ingroup to obtain an outgroup-rooted tree.
Finally, if you want you can also perform a non-partition analysis by removing the option -p.
We will now infer a rooted tree using non-reversible models. Please download:
- An input DNA alignment file for 5 ingroup species (Yak, Cow, Goat, Sheep and Tibetan antelope). This is a sub-alignment of the alignment above. We can re-use the same partition file.
To speed up the analysis, we will perform two steps. The first step is the same as the run above to infer an unrooted tree using reversible models:
iqtree2 -s bovidae.phy -p bovidae.nex -B 1000 -T AUTO --prefix rev_dna
This run will also write the best partitioning scheme to rev_dna.best_scheme.nex file.
In the second step, we will re-use this best scheme but replace the substitution model
with the most general non-reversible DNA model, 12.12 or UNREST
(see this doc) to obtain a rooted tree:
iqtree2 -s bovidae.phy -p rev_dna.best_scheme.nex --model-joint 12.12 -B 1000 -T AUTO --prefix nonrev_dna
The option --model-joint 12.12 tells IQ-TREE use a linked substitution model 12.12 across
all partitions. This is to avoid potential over-parameterization as this is very
parameter-rich model with 12 parameters.
The resulting tree extracted from .iqtree file might look like this:
NOTE: Tree is ROOTED at virtual root '__root__'
Numbers in parentheses are ultrafast bootstrap support (%)
+---Yak
+------| (72)
| | +----------Goat
| | +---| (100)
| | | +----------Sheep
| +-----------------------------------| (95)
| +-------------Tibetan_antelope
|
+**Cow
|
+**__root__
(You can better visualize the .treefile in a tree viewer software).
This run will write an additional tree file nonrev_dna.rootstrap.nex with rootstrap
support values (see box below for definition) annotated on every branch of the tree. If you open this file in FigTree
it may look like this (click on "Branch Labels" and choose rootstrap for "Display"
as shown in the figure):
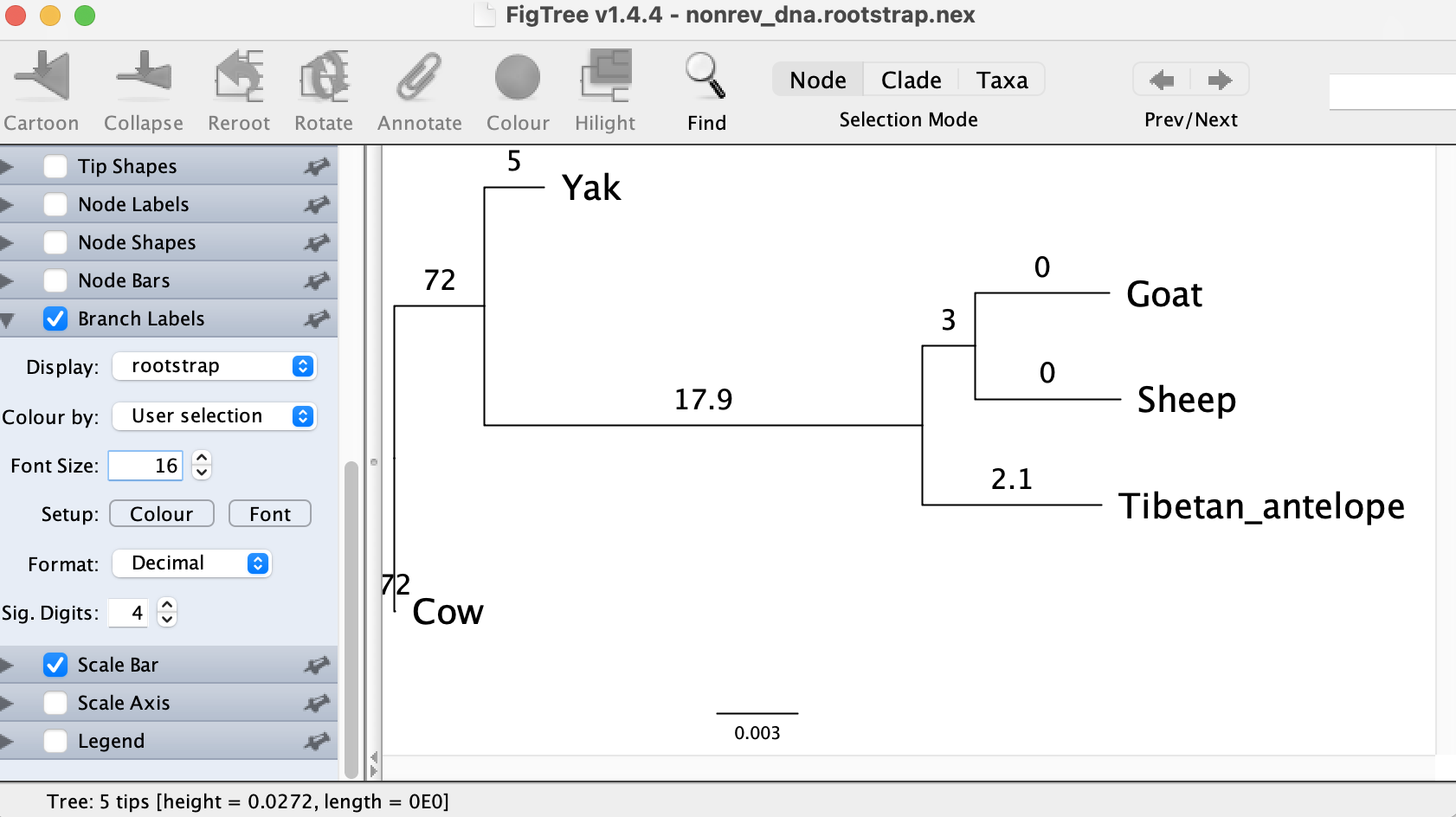
It shows that the tree might be rooted in the branch leading to Cow
with a rootstrap support of 72%, which is rather low. The 2nd best
branch separating Cow and Yak from the rest has a rootstrap support of 17.9%. So with this dataset the DNA model cannot reliably tell where
the root position is, but at least provides some candidates.
Rootstrap: To compute rootstrap supports, we conduct a bootstrap analysis to obtain a number of rooted bootstrap trees using non-reversible models. We define the rootstrap support for each branch in the maximum likelihood (ML) tree, as the proportion of rooted bootstrap trees that have the root on that branch. The rootstrap support values are computed for all the branches including external branches. The sum of the rootstrap support values along the tree are always smaller than or equal to one. A sum that is smaller than one can occur when one or more bootstrap replicates are rooted on a branch that does not occur in the ML tree.
We will now try the amino-acid model to see if that helps. We again use -st NT2AA
option to conveniently perform this analysis:
# step 1: infer unrooted tree with reversible models
iqtree2 -s bovidae.phy -p bovidae.nex -B 1000 -T AUTO -st NT2AA --prefix rev_aa
# step 2: infer rooted tree with linked non-reversible models
iqtree2 -s bovidae.phy -p rev_aa.best_scheme.nex --model-joint NONREV -B 1000 -T AUTO -st NT2AA --prefix nonrev_aa
The option --model-joint NONREV tells IQ-TREE to use the most general amino-acid model
NONREV and to link the NONREV model parameters across all partitions: NONREV has
379 parameters and linking them across partitions will avoid over-parameterization.
The tree extracted from nonrev_aa.iqtree file now may look like:
NOTE: Tree is ROOTED at virtual root '__root__'
Numbers in parentheses are ultrafast bootstrap support (%)
+------------Yak
+--------------------------------| (100)
| +----------------Cow
|
| +-------------------Goat
| +--------| (100)
| | +---------------------Sheep
+--------------------------| (100)
| +-----------------------------Tibetan_antelope
|
+**__root__
Interestingly, the amino-acid model suggests a different root position compared with the DNA model. But this position agrees with the outgroup rooting approach. And the tree nonrev_aa.rootstrap.nex with rootstrap supports look like:
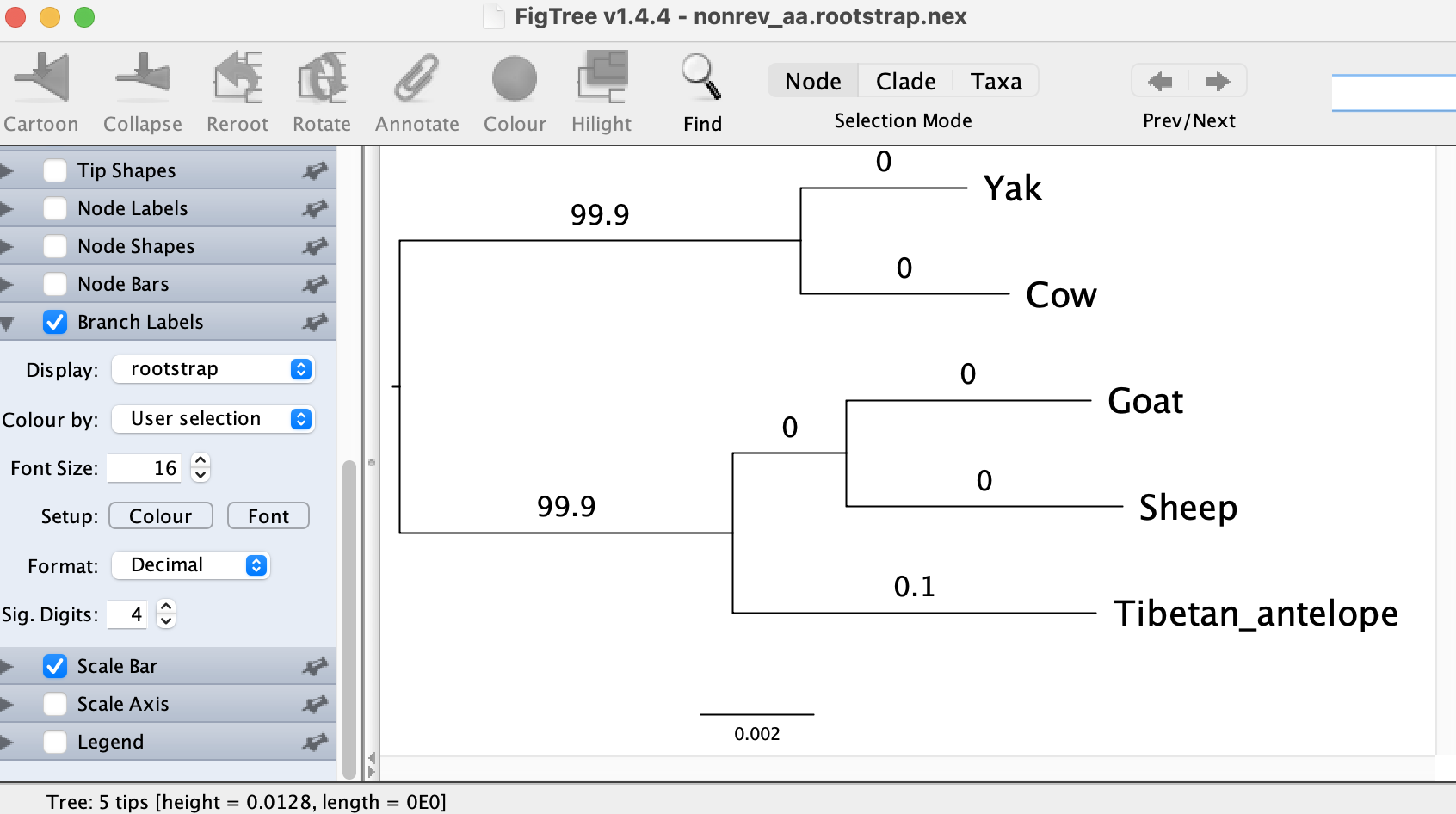
That means, the branch separating Yak and Cow from the rest receives a very high rootstrap support of 99.9%. Therefore, the amino-acid model seems to have a much higher power to detect the root, compared with the DNA model.
The rootstrap introduced above is one way to measure our confidence in the root placement, but it is not a
statistical test. Alternatively, we can apply the tree topology tests to compare the log-likelihoods of the trees being rooted
on every branch of the ML tree. IQ-TREE v2.1.3 provides a convenient option --root-test
that will re-root the tree on every branch and perform the test for you. So you can run:
iqtree2 -s bovidae.phy -p rev_aa.best_scheme.nex --model-joint NONREV -st NT2AA --root-test -zb 1000 -au -te nonrev_aa.treefile --prefix nonrev_aa_test
-zb 1000 -au is to perform several tree topology tests including the approximately-unbiased (AU) test for the tree found above (-te nonrev_aa.treefile). This run will
write a file nonrev_aa_test.roottest.csv which might look like:
# Test results for rooting positions on every branch
# This file can be read in MS Excel or in R with command:
# dat=read.csv('nonrev_aa_test.roottest.csv',comment.char='#')
# Columns are comma-separated with following meanings:
# ID: Branch ID
# logL: Log-likelihood of the tree rooted at this branch
# deltaL: logL difference from the maximal logl
# bp-RELL: bootstrap proportion using RELL method (Kishino et al. 1990)
# p-KH: p-value of one sided Kishino-Hasegawa test (1989)
# p-SH: p-value of Shimodaira-Hasegawa test (2000)
# c-ELW: Expected Likelihood Weight (Strimmer & Rambaut 2002)
# p-AU: p-value of approximately unbiased (AU) test (Shimodaira, 2002)
ID,logL,deltaL,bp-RELL,p-KH,p-SH,c-ELW,p-AU
1,-90388.66044,0,0.983,0.96,1,0.9695602131,0.9975595105
8,-90401.6833,13.02286164,0.005,0.04,0.19,0.01262108065,0.00558089101
5,-90401.68371,13.0232665,0.01,0.04,0.19,0.01262245766,0.006374455939
3,-90410.10499,21.44455589,0.002,0.016,0.104,0.002397842346,0.001014359781
2,-90410.1084,21.44796542,0,0.016,0.104,0.002389013519,0.0008999725939
6,-90413.04441,24.38397245,0,0.005,0.059,0.0002047272296,0.0004975092439
7,-90413.04797,24.38753181,0,0.005,0.059,0.0002046654974,0.0005061888325
The branches are sorted by log-likelihoods in descending order. The last column (p-AU)
shows the p-values of the AU test. The branch ID 1 has an AU p-value of 0.9975595105,
whereas all other branches has p-values < 0.01. To associate branch ID you can return to the FigTree window for nonrev_aa.rootstrap.nex file and select "Display" to "id" in the "Branch Labels" tab.
The conclusion from this analysis: we can reject all rooting positions on branches other than branch ID 1, which agrees with the rootstrap measure.
TIP: These options --root-test -zb 1000 -au can be combined with the rootstrap run
in the previous section to calculate the rootstrap support values and the rooting test p-values in one single analysis.