{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ARDaP was written by Derek Sarovich (@DerekSarovich), with database construction, code testing and feature design by Danielle Madden (@dmadden9), Eike Steinig (@EikeSteinig) (Australian Institute of Tropical Health and Medicine, Australia) and Erin Price (@Dr_ErinPrice).
- Introduction
- Installation
- Resource Managers
- ARDaP Workflow
- Usage
- Parameters
- Inclusion of Assembled Genomes
- Custom Database Creation
- AMR Report Summary
- Troubleshooting
- Citation
ARDaP (Antimicrobial Resistance Detection and Prediction) is a pipeline designed to identify genetic variants (i.e. single-nucleotide polymorphisms [SNPs], insertions/deletions [indels], copy-number variants [CNVs], and gene loss) associated with antimicrobial resistance (AMR) from microbial (meta)genomes or (meta)transcriptomes. Further, ARDaP reports these variants in a user-friendly manner that does not require extensive domain-specific knowledge, and that links AMR genotype to AMR phenotype. The impetus behind developing ARDaP was our frustration with current methodology being unable to detect AMR conferred by "complex" chromosomal alterations, and accurate AMR detection from mixtures, meaning that many tools cannot offer comprehensive AMR determinant detection. Our species of interest, Burkholderia pseudomallei, Pseudomonas aeruginosa, and Haemophilus influenzae*, can develop AMR in a multiple ways, predominantly through chromosomal gene loss, CNVs, SNPs, and indels; and in B. pseudomallei, gene gain plays no role in conferring AMR, rendering many existing AMR tools entirely ineffective. ARDaP first identifies all genetic variation in a microbial sequence data (either .fasta assemblies or Illumina paired-end data; other data types currently not supported), and then interrogates this information against a user-created database of AMR determinants. The software will then summarise the identified AMR determinants and produce an easy-to-interpret summary report.
*H. influenzae module still under development
Github + Conda
- Download the latest installation with git clone. Recommended to place this installation in a bin folder or other easily accessible location.
git clone https://github.com/dsarov/ardap.git ./ardap
- Install an ardap environment using conda
conda env create --name ardap -f ./ardap/env.yaml
or use the full environment if you are having issues with version clashes
conda env create --name ardap -f ./ardap/env_full.yaml
Load the conda environment
conda activate ardap
- Install Resfinder dependencies.
pip3 install tabulate biopython cgecore gitpython python-dateutil
The pipeline itself is either run by calling the main.nf or from a local respostory in a local nextflow cache.
Load the conda environment:
conda activate ardap
And run from a local installation:
nextflow run ~/bin/ardap/main.nf
or nextflow cache:
nextflow run dsarov/ardap
The local cache can be updated with:
nextflow pull dsarov/ardap
If you want to make changes to the default nextflow.config file, clone the workflow into a local directory and change parameters in nextflow.config:
nextflow clone dsarov/ardap install_dir/
Or navigate to the nextflow install path of ARDaP and change the nextflow.config in that location.
ARDaP is implemented in Nextflow. More information about Nextflow can be found here
ARDaP can be called from the command line through Nextflow. This will pull the current workflow into local storage. Any parameter in the configuration file nextflow.config can be changed on the command line via -- dashes, while Nextflow runtime parameters can be changed via - dash.
For example, to run Nextflow with the default cluster job submission template profile for PBS, and activate the mixture setting in ARDaP, we can run:
nextflow run dsarov/ardap --executor pbs --mixtures
or
nextflow run /path/to/ardap/main.nf --executor pbs --mixtures
The nextflow pipeline language allows different error strategies to be applied to make sure your pipeline runs as seamlessly as possible. ARDaP will attempt to re-run any failed jobs four times before giving up. Frequent crashes may represent a bug and should be reported, but an occasional crash may happen and should be mostly mitigated by the "retry" error strategy.
ARDaP is written in the Nextflow language, and as such, has support for most resource management systems.
The list of schedulers and default template profiles is in nextflow.config and can be selected when the pipeline is initiated with the --executor flag. For example, if you want to run ARDaP on a system with PBS, simply set --executor pbs when initialising ARDaP. Most popular resource managers are supported (e.g. sge, slurm). The default configuration is to run on the local system without a scheduler.
If you require more information about how to set your resource manager (e.g. memory, queue, account settings), please see https://www.nextflow.io/docs/latest/executor.html
If you would like to just submit jobs to the resource manager queue without monitoring, then use of the screen or nohup commands will allow you to run the pipeline process in the background, and won't kill the pipeline if the shell is terminated. Examples of nohup are included below.
If you are running the pipeline on a system with overly draconian resource restrictions, or you need to specify specific queues because the default queue has a time limit of one hour, additional variables can be added to the nextflow.config file using the “clusterOptions” assignment.
e.g. clusterOptions = "-A account_name" clusterOptions = "--partition=general"
To achieve high-quality variant calls, ARDaP incorporates the following programs into its workflow:
- ART
- Trimmomatic
- Seqtk
- Burrows Wheeler Aligner (BWA)
- SAMTools
- Picard
- Genome Analysis Toolkit (GATK)
- BEDTools
- Pindel
- Mosdepth
- SNPEff
- Resfinder
- SQLite
- FastTree 2
Optional Parameter:
--mixtures Optionally perform within-species mixture analysis (e.g. from metagenomic/metatranscriptomic data) for a particular species of interest. Run ARDaP with the --mixtures flag for analysis with multiple strains and/or meta-omic data. Default=false
Example:
$ nextflow run dsarov/ardap --species Burkholderia_pseudomallei --mixtures
or
$ nextflow run /path/to/ardap/main.nf --species Pseudomonas_aeruginosa --mixtures
--size ARDaP can optionally down-sample your read data to run through the pipeline more quickly (integer value expected). Default=1000000, which corresponds to ~50x coverage for a 6Mbp genome. To switch down-sampling off, specify --size 0. When mixture analysis is requested, this option should be switched to 0 for the most sensitive ARM determinant detection.
Example:
$ nextflow run dsarov/ardap --species Pseudomonas_aeruginosa --size 0 --mixtures
or
$ nextflow run /path/to/ardap/main.nf --species Pseudomonas_aeruginosa --size 0 --mixtures
--phylogeny Use this flag if you would like a whole-genome phylogeny, or an annotated variant matrix file. Note that this may take a long time if you have a large number of isolates. Default=false
Example:
$ nextflow run dsarov/ardap --species Pseudomonas_aeruginosa --phylogeny
or
$ nextflow run /path/to/ardap/main.nf --species Burkholderia_pseudomallei --phylogeny
--species Use this flag to specify an ARDaP database that contains species-specific AMR determinant information.
Currently there are ARDaP databases available for:
Pseudomonas aeruginosa --species Pseudomonas_aeruginosa
Burkholderia pseudomallei --species Burkholderia_pseudomallei
For example:
nextflow run dsarov/ardap --species Pseudomonas_aeruginosa
or
$ nextflow run /path/to/ardap/main.nf --species Pseudomonas_aeruginosa
If you don't want to constantly use the flags for different species, this setting can be changed in the nextflow.config file.
--fastq ARDaP, by default, expects reads to be paired-end, Illumina data in the following format:
STRAIN_1.fastq.gz (first pair)
STRAIN_2.fastq.gz (second pair)
Reads not in this format will be ignored unless you change the --fastq flag to match the read naming on your system.
Example:
If your reads are in the following format
STRAIN_1_sequence.fq.gz (first pair)
STRAIN_2_sequence.fq.gz (second pair)
nextflow run dsarov/ardap --fastq "*_{1,2}_sequence.fq.gz"
or
$ nextflow run /path/to/ardap/main.nf --fastq "*_{1,2}_sequence.fq.gz"
Please make sure to include the quotation marks when specifying the fastq format eg "*_{1,2}_sequence.fq.gz" not *_{1,2}_sequence.fq.gz
Although it is strongly recommended to use raw Illumina reads to enable to most robust AMR detection (especially CNVs and gene upregulation), ARDaP can optionally include assembled genomes in the workflow using the --assemblies flag. To do so, please include all genome assemblies in an "assemblies" subdirectory of the main analysis directory. These files will need to be in FASTA format with the .fasta extension. ARDaP will automatically scan for the subdirectory "assemblies" and include all files identified with a .fasta extension. Synthetic reads will be synthesised using ART and included in all downstream analysis. Currently there is a limitation that at least some strains must have short-read data in addition to the assembled genomes. This is a known limitation but should be addressed in a future update.
When creating your own custom database, please use the database templates to ensure ARDaP compatibility.
ARDaP's creators have, to date, developed and validated a custom AMR database for Burkholderia pseudomallei, and a Pseudomonas aeruginosa AMR database is in progress; however, ARDaP is capable of detecting and predicting AMR determinants in any microbial species of interest. Adding species requires populating a database of all known AMR determinants acquired from: a) chromosomal mutations (SNPs, indels, CNVs, and gene loss); and b) horizontal gene gain (through incorporation and improved annotation of the CARD database). To develop a truly comprehensive AMR database is a nontrivial and ongoing effort; however, doing so is essential for enhancing the value of tools like ARDaP for AMR determinant identification from microbial genomes. It is recommended that a thorough literature review first be carried out, followed by manual cataloguing and verification of individual AMR determinants that specify not only what class, but what specific antibiotic/s are affected by the presence of an AMR variant.
The species-specific databases are incorporated into ARDaP using SQLite. ARDaP looks for information in specific tables of your SQLite database, and the structure and names of columns in these tables must not be modified when creating the custom database; if they are, ARDaP will be unable to properly detect and annotate the AMR information in a given strain.
The following sections outline, with an example, what components of the database can be customised to tailor the database for any microbial species of interest. The ARDaP tables include:
- A table specifying the clinically-relevant antibiotics for the bacterial species of interest. Required columns include: Antibiotic, abbreviation, drug class, drug family
- A Coverage table (for identifying CNVs or gene loss that confer AMR). Required columns include: Gene, Locus tag, chromosome, start coordinates, end coordinates, upregulation or loss, antibiotic affected, known combination, comments.
- A table for the genome-wide association study (GWAS) used in the predictive component of ARDaP. Note that incorporation of this table requires a large amount of genomes, with accompanying AMR phenotypic data from sensitive and resistant strains from the microbial species of interest. Depending on the target microbe, this table may not be workable; for example, too few genomes are currently available for a GWAS to be undertaken in the B. pseudomallei module of ARDaP. In contrast, there are sufficient public genomes with accompanying AMR profiles in the P. aeruginosa module for a GWAS approach to be used. Required columns include: GWAS ID, genomic coordinate, reference base, mutation type, specific ‘antibiotic’ resistant p-value and specific ‘antibiotic’ intermediate resistance p-value (note: these resistant and intermediate p-values need to be included for every individual antibiotic)
- A table of mutational variants (i.e SNPs and indels) for conferring AMR. Required columns include: Gene name, variant annotation, alternative variant annotation, antibiotic affected, known combination, comments
NB. A complete, closed archetypal reference genome should be used where possible, and this genome must be present in the SnpEff database. Run the following command to identify the available SnpEff databases for your microbe of interest: e.g. conda activate ardap && snpEff databases | grep 'Burkholderia_pseudomallei'
Populating the SQLite AMR database
Important: DO NOT alter any information in the 'Structure' tabs; otherwise ARDaP will not work!
A. Antimicrobial list
The ‘Data’ tab located within the 'Antibiotics' table (Figure 1) enables the database creator to add new rows listing all relevant antimicrobials, along with information about antimicrobial drug class and family. The abbreviations in this 'Data' table link to the final AMR reports generated by ARDaP. In other words, ARDaP will only report AMR determinants when the antimicrobial abbreviations have been added to this tab. Please use standardised antimicrobial abbreviations. Make ensure you commit changes when new additions/changes are made by clicking on the green checkbox in SQLite.
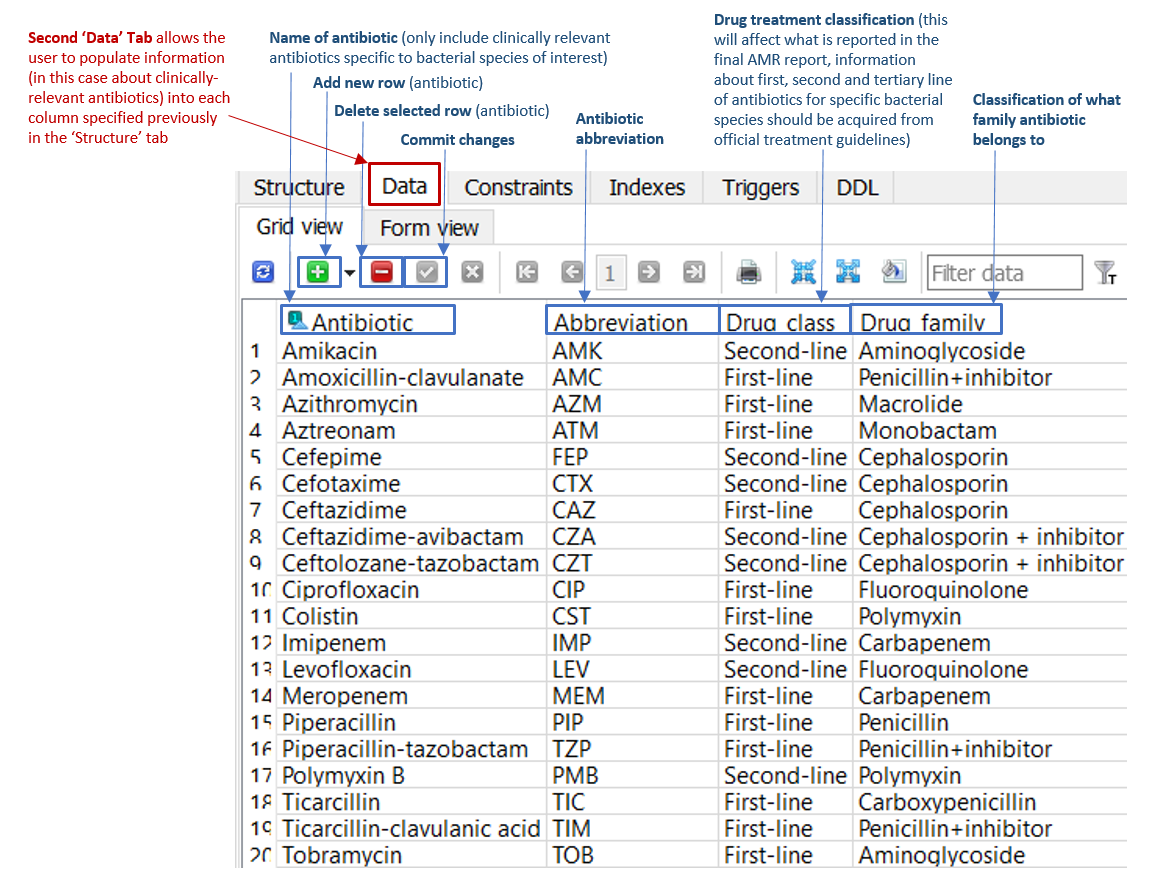 Figure 1: 'Data' tab of the 'Antibiotics' table in the P. aeruginosa ARDaP SQLite database
Figure 1: 'Data' tab of the 'Antibiotics' table in the P. aeruginosa ARDaP SQLite database
B. Variants table - for SNPs and indels conferring AMR
SNP and indel AMR variants can be added to the 'Data' tab of the 'Variants_SNP_indel' table (as illustrated in Figure 2). Required information includes the AMR gene name, the SnpEff AMR variant annotation (and alternate gene annotation, if applicable), and the antimicrobial/s affected by the variant. For example, in row 3 of Figure 2, a frameshift mutation is present in the P. aeruginosa ampD AMR gene at residue 18 (serine), which confers AMR towards the cephalosporin antibiotics, ceftazidime (CAZ) and cefipime (FEP). Within this table is also a ‘known combination’ column, which should be populated in instances where two or more stepwise mutations are required to confer AMR.
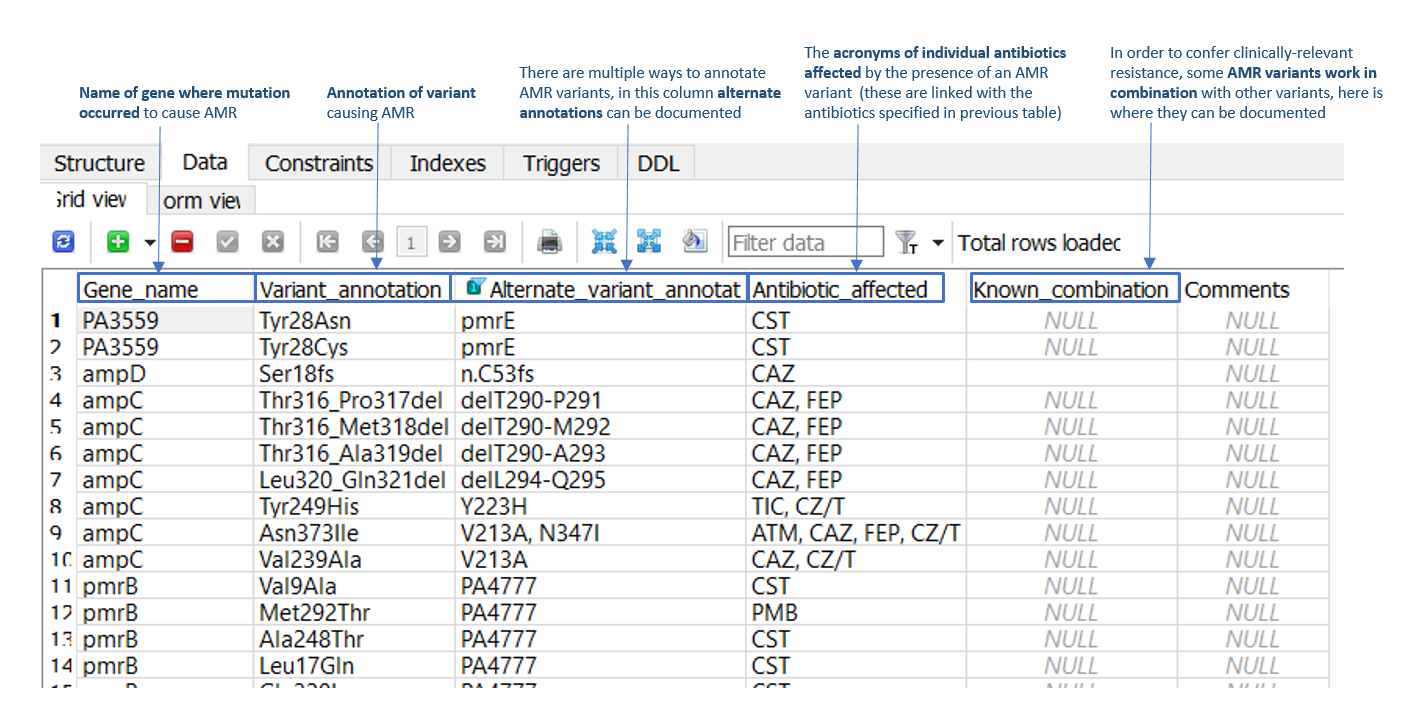 Figure 2: 'Data' tab of the 'Variants_SNP_indel' table in the P. aeruginosa ARDaP SQLite database
Figure 2: 'Data' tab of the 'Variants_SNP_indel' table in the P. aeruginosa ARDaP SQLite database
C. Coverage table - for AMR conferred by CNVs, gene loss, or gene upregulation (in RNA-seq data)
Gene loss and CNVs are important, yet underrecognised, causes of AMR. The 'Data' tab of the 'Coverage' table (e.g. Figure 3) should be used to list genes that confer AMR when lost (either fully or partially), or for genes where CNVs or upregulation cause AMR.
 Figure 3: 'Data' tab of the 'Coverage' table in the P. aeruginosa ARDaP SQLite database
Figure 3: 'Data' tab of the 'Coverage' table in the P. aeruginosa ARDaP SQLite database
ARDaP summarises the AMR findings, if any, for each (meta)genome or (meta)transcriptome of interest in an easy-to-interpret report (Figure 4). This report also correlates AMR genotype with AMR phenotype. Finally, ARDaP flags quality issues, incorrect species, and natural genetic variation that does not confer AMR.
 Figure 4: Easy-to-interpret AMR report generated by ARDaP
Figure 4: Easy-to-interpret AMR report generated by ARDaP
Q: My pipeline crashed. Where do I go to figure out what happened?
A: Nextflow (and ARDaP) will output A LOT of information about why a certain step failed and how to go about fixing the error. The first place to start looking is in the nextflow output to screen. This output will tell you where each step of the pipeline is being processed and where the log files are kept for that step.
Q: My pipeline crashed with an error 143 - Job terminated by sytem.
A: Take a look at the nextflow.config file to see if sufficient resources are being requested for the individual steps. These parameters have been chosen as a "most common use" scenario but they may not be suitable for all computational systems or all data. ARDaP will retry a number of times at each failed step but it may help to increase resources at specific steps, especially if repeated failures are noted.
Please send bug reports to [email protected] or log them in the github 'issues' tab
=======