Workflow Jaffe lab
Instructions: Here is an example of the morphological profiling data analysis workflow followed by the Carpenter lab. Please write down the workflow that you use in your own group for proteomics data analysis, which admittedly will be different from what we do in morphological profiling. Also, please provide references where relevant. During the hackathon, each group will be allotted 8 minutes to present their workflow.
Source:https://panoramaweb.org/labkey/project/LINCS/begin.view
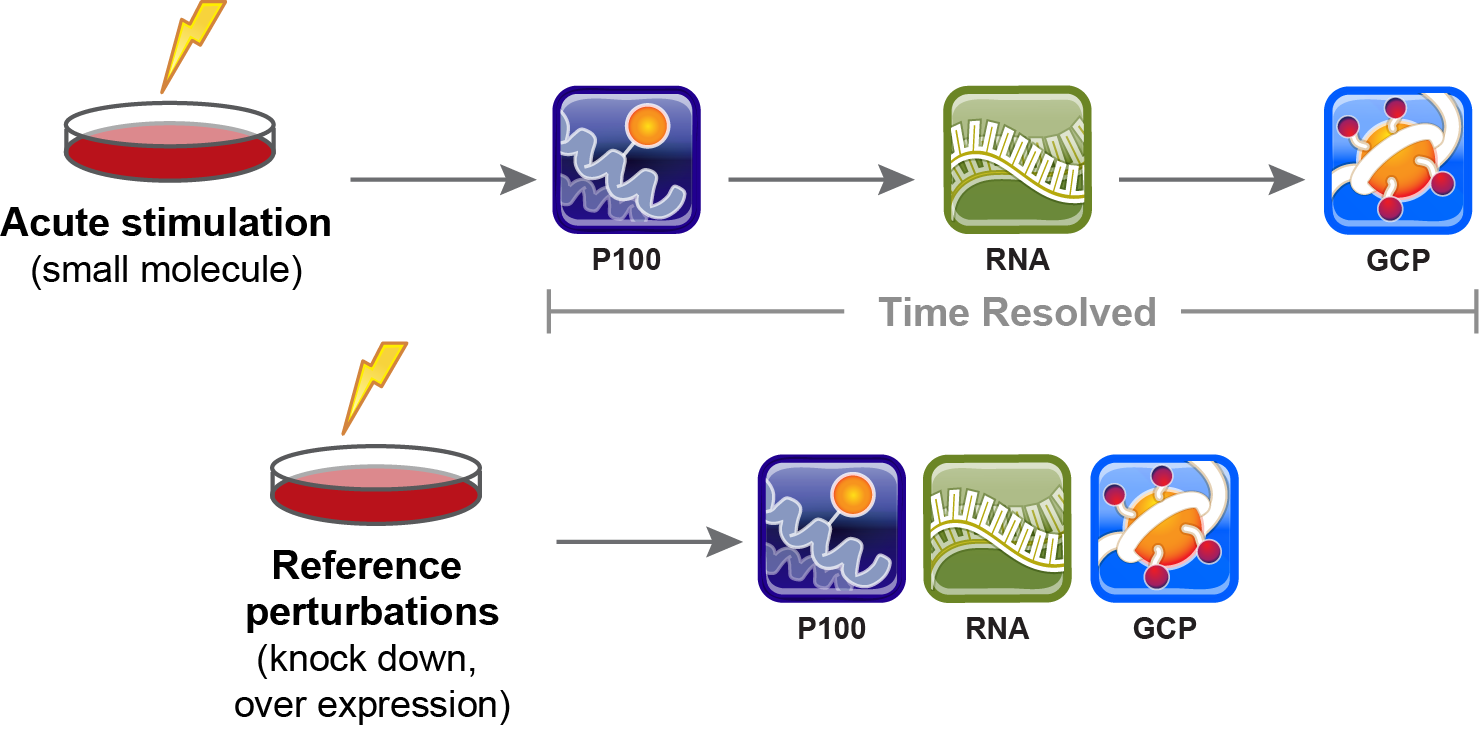
Our center makes systematic perturbations (drug treatments, gene disruption/editing, and directed differentiation) in a variety of biological models including cancer and neurodevelopment. We read out molecular changes in the cells in the spaces of phosphosignaling (P100) and histone modification (GCP). In collaboration with the LINCS Center for Transcriptomics, we also read out changes in transcription using their L1000 assay. Look here for more information about our center or download our poster here.
From: https://panoramaweb.org/labkey/project/LINCS/begin.view
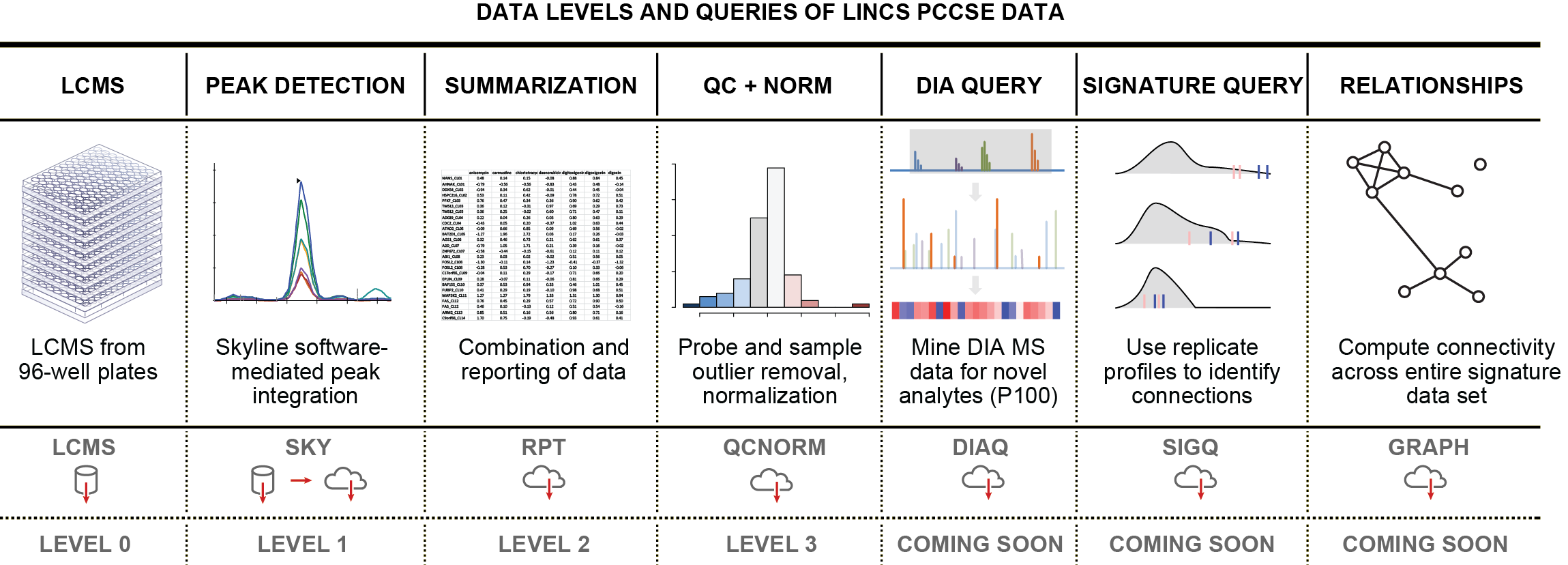
LINCS espouses the concept of making different data levels available for public use. Different data levels correspond different steps along our processing workflow. The LINCS PCCSE levels are defined as follows:
- Level 0 - Raw Mass Spectrometry Data - will be available through a chorusproject.org repository in the future
- Level 1 - Probe Reads - Curated Skyline documents; available on this website, including metadata
- Level 2 - Raw Numerical Data - Matrix data of extracted signal ratios of endogenous probes vs. internal standards; available on this website, including metadata
- Level 3 - Normalized and QC'ed Numerical Data - Matrix data derived from Level 2 after automated processing and normalization; available on this website, including metadata